Микроцитарная гемолитическая анемия относится к наследственным заболеваниям. Наследуется по аутосомно-доминантному типу. Сущность патологического процесса заключается в дефекте структуры мембраны эритроцитов. В эксперименте на мышах показано, что при наследственном микросфероцитозе отсутствует белок мембраны эритроцитов спектрин. Костный мозг при этом продуцирует неполноценные эритроциты, отличающиеся от нормальных тем, что они меньшего диаметра и имеют форму не двояковогнутой, а двояковыпуклой линзы, вследствие чего и названы микросфероцитами. При этом объем эритроцитов остается в пределах нормальных колебаний, и анемия, являясь по существу сфероцитарной, микроцитарной не является. Название же «микросфероцитарная» отражает тот факт, что за счет изменения формы и уменьшения диаметра эритроцитов в мазке крови они выглядят аналогично микроцитам. Аппаратный же анализ показывает нормальные параметры красных клеток крови.
Мембрана таких эритроцитов обладает повышенной проницаемостью для ионов натрия. Это приводит к их набуханию. Сферическая форма и особенности структуры белка мембраны эритроцитов нарушают их способность проходить узкие места кровотока без повреждений и разрушения. Основным местом гемолиза является селезенка, исследования показали, что продолжительность жизни эритроцитов у таких больных составляет 8-15 дней вместо 90-120 у здоровых людей.
Первые признаки заболевания могут проявиться в детском возрасте, но чаще — в юношеском и зрелом. В течение длительного времени единственным признаком болезни является желтушное окрашивание склер и кожи. Течение волнообразное. Причиной усиления гемолиза и, соответственно, ухудшения состояния чаще всего являются инфекция, переохлаждение, беременность. Развивается слабость, появляются одышка и учащенное сердцебиение при физической нагрузке. Степень интенсивности желтухи может быть различной: от незначительной до резко выраженной. С каждым обострением желтушность усиливается. На фоне хронической интоксикации и гипоксии тканей могут наблюдаться отставание в росте и физическом развитии, снижение толерантности к физическим нагрузкам. У больных возможны скелетные аномалии в виде высоко стоящего твердого нёба («готическое нёбо») и др.
Следует иметь в виду, что течение этого вида гемолитической анемии может осложняться желчнокаменной болезнью вследствие образования желчных камней. В таких случаях возможно появление болевых приступов в правом подреберье. Интенсивность желтухи усиливается за счет присоединения обтурационного механизма. Характерным признаком заболевания является увеличение селезенки, а в дальнейшем и печени. Селезенка может увеличиваться до значительных размеров, что обусловлено усиленным гемолизом в ней эритроцитов.
В период обострения в моче повышается содержание уробилина, а в кале — стеркобилина. Содержание эритроцитов и уровень гемоглобина в период ремиссии может быть в норме, а в период усиления гемолиза развивается малокровие. Обращает на себя внимание наличие в периферической крови эритроцитов меньшего диаметра при сохранении или даже увеличении их объема. Сфероциты интенсивно окрашены и не имеют просветления в центре. Такие эритроциты обладают пониженной осмотической стойкостью. Гемолиз их может начинаться при концентрации натрия хлорида 0,60-0,70 % вместо 0,44-0,46 % в норме. Понижена и их резистентность. Как и для любой другой формы гемолитической анемии, характерно повышение количества ретикулоцитов, что свидетельствует об ускоренном их вымывании из костного мозга. Количество лейкоцитов как правило, не изменяется. В костном мозге наблюдается выраженная гиперплазия эритроидных клеток. Их количество может повышаться до 30-50% вместо 15-25% в норме.
Лечение. При стабильном течении, когда заболевание проявляется слабо выраженной желтушной окраской кожи, при хорошем самочувствии и отсутствии признаков анемии каких-либо особых методов лечения не требуется. При частых гемолитических кризах, сопровождающихся развитием анемии и осложнениями (желчнокаменная болезнь, инфаркт селезенки), показана спленэктомия. После спленэктомии исчезает желтуха, нормализуются уровень гемоглобина и количество эритроцитов, снижается количество ретикулоцитов, улучшается функция печени, содержание билирубина в сыворотке крови снижается до нормальных показателей. Однако характер эритропоэза не изменяется. Остаются микросфероцитоз и пониженная осмотическая стойкость эритроцитов. Улучшение состояния достигается лишь за счет удаления органа, где совершается наиболее интенсивный гемолиз эритроцитов, что приводит к увеличению продолжительности их жизни.
источник
 Анемия Под определение «гемолитическая анемия» подпадает любая анемия, характеризующаяся преобладанием гемолиза (процессом распада эритроцитов) над эритропоэзом (процессом образования эритроцитов). Гемолиз может быть иммунный и неиммунный. Гемолитическая анемия бывает наследственные и приобретенные.
Анемия Под определение «гемолитическая анемия» подпадает любая анемия, характеризующаяся преобладанием гемолиза (процессом распада эритроцитов) над эритропоэзом (процессом образования эритроцитов). Гемолиз может быть иммунный и неиммунный. Гемолитическая анемия бывает наследственные и приобретенные.
- Наследственная форма гемолитической анемии, обусловленная нарушением мембраны эритроцитов
- Наследственная форма гемолитической анемии, обусловленная нарушением активности ферментов эритроцитов
- Наследственная форма гемолитической анемии, обусловленная нарушением синтеза или структуры гемоглобина
- Анемия, обусловленная влиянием антител
- Анемия, обусловленная изменением структуры мембраны, вызванной соматической мутацией
- Анемия, обусловленная механическим повреждением оболочки эритроцитов
- Анемия, вызванная химическим повреждением эритроцитов
- Анемия, вызванная дефицитом витаминов (фолиевой кислоты и цианокобаламина)
- Анемия, вызванная разрушением эритроцитов паразитами
Болезнь Минковского-Шоффара (наследственный микросфероцитоз) – группа наследственных гемолитических анемий, характеризующихся образованием микросфероцитов (шаровидных эритроцитов) и обусловленных дефектом протеинов цитоскелета эритроцитов. При этом эритроциты теряют часть мембраны, уменьшается соотношение площади поверхности к объему, в результате чего эритроцит превращается в микросфероцит. Как правило, патология наследуется по аутосомно-доминантному признаку. Распространенность наследственного микросфероцитоза составляет примерно 1 случай на 1000-4500 человек.
При наследственном микросфероцитозе генетические нарушения влияют на протеины цитоскелета, преимущественно на те, которые объединяют цитоскелет с мембраной эритроцита. У большинства больных отмечается значительный дефицит спектрина, и только в некоторых случаях этот дефицит обусловлен генетическими дефектами самого спектрина.
Главные признаки наследственного микросфероцитоза – анемия, желтуха, спленомегалия (увеличенная селезенка). Анемия возникает из-за внутриклеточного распада эритроцитов. Желтуха развивается посредством непрямой гипербилирубинемии, может быть непостоянной и, как правило, слабо выражена у детей раннего возраста. Повышенное содержание билирубина в желчи часто является причиной образования пигментных желчных камней (даже у детей). Увеличение селезенки (спленомегалия) отмечается практически во всех случаях. При системных инфекционных патологиях интенсивность гемолиза может увеличиваться, в результате чего развивается спленомегалия.
Тяжелые формы наследственного микросфероцитоза характеризуются деформацией скелета: изменение расположения зубов, акрокефалия (башенный череп), высокое верхнее небо, микрофтальмия (уменьшение глазного яблока). В некоторых случаях отмечаются укороченные мизинцы. Могут образовываться трофические язвы на ногах.
Наследственный микросфероцитоз сопровождается апластическими кризами, которые провоцируются инфекцией (особенно парвовирусной).
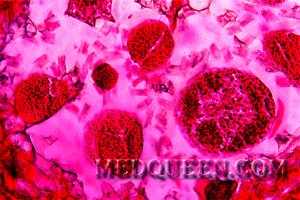
Микросфероцитоз – характерное изменение формы эритроцитов при этой патологии. При анализе мазка крови в биологическом материале наблюдаются микросфероциты в виде мелких клеток без центрального просветления (см рисунок 1). Отметим, что обнаружение микросфероцитов в мазках не всегда является признаком наследственного сфероцитоза.
Рисунок 1. Наследственный микросфероцитоз. Микросфероциты в мазке периферической крови (окр. по Романовскому-Гимзе, ув. ×100)
Такой признак обнаруживается при аутоиммунной гемолитической анемии с неполными тепловыми агглютинами, при наследственных дизэритропоэтической анемии. Средний объем эритроцитов, как правило, остается в норме или незначительно снижен. Показатель среднего содержания гемоглобина в эритроцитах в норме или незначительно повышен. Средняя концентрация гемоглобина в эритроцитах повышена почти у 50% пациентов.
Количественным показателем сферичности эритроцитов является осмотическая устойчивость (она снижена). Уровень ретикулоцитов в крови при гемолитическом кризе может значительно повышаться. Миелограмма показывает резкое раздражение красного ростка. Дифференциальный диагноз проводят с аутоиммунной гемолитической анемией, для которой характерна положительная проба Кумбса, отсутствие этой патологии среди родственников пациента и отсутствие данных о начале заболевания в детском возрасте.
Основной метод лечения анемии при наследственном микросфероцитозе – спленэктомия, с помощью которой устраняется анемия; при этом нельзя устранить морфологический дефект эритроцитов.
Наследственная гемолитическая анемия, обусловленная дефицитом глюкозо-6-фосфат дегидрогеназы эритроцитов – наиболее распространенная ферментопатия эритроцитов из группы ферментопатий пентозофосфатного пути метаболизма глюкозы. Глюкозо-6-фосфатдегидрогеназа эритроцитов – олигомер (в зависимости от условий может быть димер или тетрамер), который состоит из субъединиц с молекулярной массой 56 000 D. По данным ВОЗ (Всемирной организации здравоохранения) во всем мире количество людей, страдающих этой патологией, составляет более 200 млн. Наиболее широкое распространение этого заболевания характерно для Средиземноморского региона (Сицилия, Греция, Сардиния), негроидной расы, жителей Ближнего и Дальнего востока.
Клиническая картина при наследственной форме гемолитической анемии полиморфна: степень тяжести патологии может колебаться от гемолитической анемии, возникающей спонтанно после рождения, до гемолитических кризов. Гемолитический криз, который может провоцироваться метаболическим ацидозом или гипогликемией, развивается за несколько часов. В тяжелых случаях у больного развивается гемоглобинурия и шок. Также наблюдаются желтуха, моча приобретает бурый или черный цвет, одышка, диарея, рвота, снижение артериального давления, развивается тяжелая анемия, увеличиваются печень (гепатомегалия) и селезенка (спленомегалия).
Тяжелый гемолитический криз может спровоцировать развитие ДВС-синдрома (диссеминированного внутрисосудистого свертывания крови). Некоторые пациенты не переносят конские бобы (Viciafaba), после употребления которых происходит молниеносное развитие гемолитического криза (это явление также известно, как фовизм или примахиновая анемия).
Дефицит глюкозо-6-фосфат дегидрогеназы эритроцитов необходимо подозревать во всех случаях острого гемолиза, особенно у лиц негроидной расы и жителей средиземноморского региона. Диагноз подтверждается путем проведения лабораторных анализов. Острый гемолиз характеризуется быстрым снижением гематокрита с одновременным повышением уровня гемоглобина и непрямого гемоглобина, а также снижением уровня гаптоглобина. Анализ мазка крови показывает наличие фрагментов эритроцитов. Основой диагностики считается качественное (при необходимости – количественное) определение активности глюкозо-6-фосфат дегидрогеназы эритроцитов. У пациентов с вариантом «А-» явление аномального гемолиза проходит, как правило, самостоятельно – такие больные не нуждаются в специальном лечении. В случае развития тяжелого гемолитического криза необходимо проводить форсированный диурез, профилактику ДВС-синдрома, плазмаферез (с целью удаления продуктов гемолиза).
В случае возникновения качественной гемоглобинопатии происходит изменение аминокислотной последовательности цепей глобина. Талассемия (количественная гемоглобинопатия) характеризуется снижением образования цепей глобина без изменения их цепей. Нужно отметить, что разница между качественной и количественной гемоглобинопатиями не абсолютна.
Талассемия (анемия Кули) – группа патологий, обусловленных генетическим нарушением синтеза одной из цепей глобина. В норме процесс синтеза глобиновых цепей сбалансирован, поэтому свободных цепей глобина нет. В случае нарушения синтеза одной из цепей глобина баланс нарушается, образуются лишние цепи, которые агрегируют и откладываются в эритрокариоцитах. Среди жителей Средиземноморья наиболее распространена β-талассемия.
«Большая талассемия» (болезнь Кули, β-талассемия) – наследственная гемолитическая анемия, впервые описанная американскими педиатрами-гематологами Томасом Бентоном Кули (Thomas Benton Cooley) и Ли (P. Lee) в статье «Серия случаев спленомегалии у детей с анемией и необычными изменениями костей» («A Series of Cases of Splenomegaly in Children, with Anemia and Peculiar Bone Changes»), где были приведены случаи у выходцев из стран Средиземноморья. Для анемии Кули характерна тяжелая степень течения с самого детства, задержка роста и изменения костей в результате увеличения объема костного мозга, возникающие в случае отсутствия соответствующего лечения). Также при этой патологии у больного наблюдаются гепатомегалия, спленомегалия, гиперспленизм, деформации черепа (монголоидное лицо, башенный череп); желтуха, бледность и отложение меланина придают коже особый медный оттенок. Кроме этого, наблюдается перегрузка железом сердца, легких, печени, поджелудочной железы и других органов эндокринной системы, переломы костей, сдавления периферических нервов, разного рода инфекционные осложнения.
Результаты лабораторных исследований периферической крови показывают гипохромную анемию, ретикулоцитоз, мишеневидные эритроциты (см рис 2-4).
Рисунок 02. Анемия Кули (большая талассемия). Периферическая кровь. Микроцитоз, выраженная гипохромия, мишеневидные нормобласты и эритроциты (окр. по Романовскому-Гимзе, ув. ×100)
Рисунок 03. Анемия Кули (большая талассемия). Периферическая кровь (окр. по Романовскому-Гимзе, ув. ×50)
Рисунок 04. Анемия Кули (большая талассемия). Периферическая кровь. Множественные мишеневидные эритроциты (окр. по Романовскому-Гимзе, ув. ×100)
Миелограмма демонстрирует раздражение «красного ростка» и повышение количества сидеробластов. Также наблюдается повышение осмотической резистентности эритроцитов и количества билирубина за счет непрямой фракции. В крови повышается содержание железа и ферритина, развивается гемосидероз (чрезмерное отложение гемосидерина в тканях) внутренних органов. При гомозиготной β-талассемии необходимо проводить пренатальную диагностику – забор клеток плода из амниотической жидкости на предмет выявления мутации генов, отвечающих за кодирование β-цепи глобина, с применением метода полимеразной цепной реакции.
Без соответствующего лечения больные анемией Кули умирают в детском возрасте. Продлить жизнь, предупредить деформации костей и задержку роста можно путем регулярных трансфузий эритроцитарной массы (лучше переливать отмытые или размороженные эритроциты) при условии поддержания достаточно высокого уровня гемоглобина. В случае значительной спленомегалии и явлениях гиперспленизма больному показана спленэктомия (удаление селезенки). С целью предотвращения развития гемосидероза пациентам периодически назначают Деферазирокс (Эксиджад) или Дефероксамин (Десферал). Излечение возможно при аллогенной трансплантации костного мозга.
Серповидноклеточная анемия обусловлена носительством гемоглобина, который меняет свою структуру в условиях гипоксии. Самой распространенной аномалией структуры гемоглобина является гемоглобинопатия Sα2β26 глу+вал. При гомозиготном носительстве можно говорить о серповидноклеточной анемии; при гетерозиготном носительстве – серповидноклеточная аномалия. Патология наследуется по аутосомно-доминантному признаку. При серповидноклеточной анемии наблюдается мутация, в результате которой в цепи глобина глутаминовая кислота заменяется валином. В результате растворимость гемоглобина S при отдаче кислорода снижается, что приводит к образованию геля.
Серповидноклеточная анемия наиболее распространена среди населения Центральной Африки, Турции, Индии, Кубы. У больных диагностируется анемия, тромботические осложнения, поражения костей и суставов (отмечаются некрозы плечевой и бедренной костей). Кроме этого, тромбозы осложняются инфарктами (сердца, легких, почек, селезенки, головного мозга), приступами сильной боли в области живота. У детей отмечаются нарушения физического (отставание в росте) и полового развития, ночное недержание мочи, нарушение зрения (тромбозы сосудов сетчатки). Также могут развиваться гемолитический, апластический и секвестрационные кризы, при этом в селезенке происходит резкое накопление эритроцитов, что вызывает гиповолемический шок и резкое снижение уровня гемоглобина.
Для анализов крови при апластической анемии характерны низкий уровень гемоглобина, наличие серповидных эритроцитов (рисунок 5), базофильная пунктация эритроцитов, их мишеневидность, повышение уровня ретикулоцитов и непрямого билирубина. Миелограмма демонстрирует раздражение «красного ростка».
Рисунок 5. Серповидноклеточная анемия. Периферическая кровь. Серповидные и мишеневидные эритроциты. выраженная гипохромия эритроцитов (окр. по Романовскому-Гимзе, ув. ×100)
В качестве лечения применяют адекватную инфузионную терапию, переливания эритроцитарной массы, оксигенотерапии.
К приобретенным гемолитическим анемиям относится группа заболеваний разного патогенеза, которые объединяет внутрисосудистый гемолиз (гемолиз эритроцитов в периферической крови). В зависимости от механизма эритролиза приобретенная гемолитическая анемия может носить иммунный и неиммунный характер. Но, несмотря на разные патогенетические механизмы, клинические признаки этих анемий часто совпадают.
Гемолитическая анемия у пациентов с протезированными клапанами сердца и сосудами развивается примерно в 10% случаев при протезированном аортальном клапане. При использовании стеллитовых запирательных элементов частота гемолиза незначительно увеличивается (по сравнению с селиконовыми). Также некоторое увеличение частоты гемолиза отмечается при наличии околоклапанной регургитации и при малом диаметре клапана. Биопротезы (свиные клапаны) в редких случаях являются причиной механического гемолиза. Гораздо реже причиной гемолиза может быть также протезированный митральный клапан, так как трансклапанный градиент давления в этом случае ниже.
Гемолиз протезированными клапанами происходит в результате одновременного действия сразу нескольких факторов:
- Значительная сила сдвига, которая при турбулентном токе крови действует на мембрану эритроцитов, особенно когда под высоким давлением кровь проходит через маленькое отверстие (например, при околоклапанной регургитации)
- Отложения фибрина на участках неплотного прилегания кольца клапана к тканям сердца
- Прямое механическое повреждение эритроцитов при закрытии запирательного элемента
Значительное разрушение эритроцитов может наблюдаться после закрытия дефекта межпредсердной перегородки типа ostium primum заплатой из синтетического материала. Умеренное сокращение жизни эритроцитов с легкой анемией или без нее может наблюдаться при значительном обызвествлении аортального клапана. Механический гемолиз обнаруживается также у пациентов, перенесших аортокоронарное и аортобедренное шунтирование.
Тяжелые случаи механического гемолиза сопровождаются тяжелой анемией, ретикулоцитозом, обнаруживаются фрагментированные эритроциты (шизоциты), гемоглобинемия и гемоглобинурия, повышается активность лактатдегидрогеназы, снижается уровень гаптоглобина. Выведение железа из организма с мочой в виде гемосидерина или гемоглобина может вызвать дефицит железа в организме. В случае развития дефицита железа пациенту назначается пероральный прием препаратов железа. Терапия препаратами железа способствует повышению уровня гемоглобина и способствует снижению сердечного выброса и снижению интенсивности гемолиза. Отметим, что ограничение физической активности также способствуют снижению интенсивности распада эритроцитов. Если предпринимаемые меры не приводят к желаемому результату, нужно полностью устранить околоклапанную регургитацию или заменить протез.
источник
· сильные боли в животе, преимущественно в области печени и левого подреберья;
· тошнота, рвота, исчезновение аппетита;
· учащение стула (непостоянный признак);
· головная боль, головокружение;
· появление судорог (грозный симптом, свидетельствующий о тяжелом состоянии больного);
· усиление бледности кожи и видимых слизистых оболочек, восковидный оттенок кожи, возможно усиление желтухи (кожа приобретает шафраново-желтый цвет);
· повышение температуры тела;
· усиление темного цвета мочи;
· увеличение селезенки и печени; селезенка болезненная в связи с периспленитом;
· усиление выраженности анемии и ретикулоцитоза при исследовании периферической крови;
· выраженный лейкоцитоз и сдвиг лейкоцитарной формулы влево;
Степени тяжести наследственной микросфероцитарной анемии
Легкая форма заболевания наблюдается приблизительно у 25% больных, характеризуется удовлетворительным состоянием, анемии нет, может наблюдаться незначительная желтушностъ кожи и видимых слизистых оболочек (по образному выражению Шаффара «больные более желтушны, чем больны»), спленомегалия выражена незначительно. Гемолиз распознается преимущественно лабораторными методами.
Средняя степень тяжести характеризуется легкой или умеренно выраженной анемией, эпизодами желтухи, выраженной спленомегалией. Довольно часто желтуха и анемия усиливаются под влиянием интенсивной физической нагрузки или вирусной инфекции.
Тяжелая форма проявляется резко выраженной анемией (часто требуется переливание эритроцитарной массы для компенсации анемии), спленомегалией, частыми гемолитическими кризами, выраженной желтухой, значительным отставанием детей в физическом развитии, апластическими кризами.
Лабораторные данные и инструментальные исследования при наследственной микросфероцитарной анемии
Общий анализ крови — нормохромная анемия различной степени выраженности, появление микросфероцитов (эритроцитов уменьшенного диаметра шарообразной формы без просветления в центре) и ретикулоцитов в большом количестве. Анемия резко усиливается после гемолитического и особенно гипопластического криза. Вне криза анемия умеренная, а при легком течении заболевания может отсутствовать. Количество ретикулоцитов возрастает при гемолитическом кризе, при гипопластическом кризе ретикулоцитоз отсутствует. Микросфероциты характеризуются уменьшением диаметра (средний диаметр 6-4 мкм), увеличением их толщины и шарообразной формой (рис.1). Чем тяжелее форма заболевания, тем большее количество микросфероцитов определяется в периферической крови.
Рис. 1. Кривые Прайс-Джонса в норме и при патологических состояниях
Количество лейкоцитов и тромбоцитов обычно нормальное. В период гемолитического криза наблюдается лейкоцитоз и выраженный сдвиг лейкоцитарной формулы влево. СОЭ увеличивается только в периоде обострения заболевания, особенно во время гемолитического криза.
Общий анализ мочи — определяется уробилинурия, а во время гемолитического криза — альбуминурия, микрогематурия.
Биохимический анализ крови — повышено содержание билирубина преимущественно за счет неконъюгированного билирубина, во время гемолитического криза возможно повышение активности аланиновой аминотрансфераэы, повышение содержания железа.
Осмотическая стойкость эритроцитов — отмечается снижение максимальной и минимальной осмотической стойкости эритроцитов.
В норме минимальная стойкость составляет 0,48-0,44%, максимальная — 0,40-0,36% раствора натрия хлорида. При наследственной микросфероцитарной анемии гемолиз начинается при более высокой концентрации натрия хлорида: минимальная осмотическая резистентность понижена: 0,7- 0,6%, а максимальная осмотическая резистентность повышена: 0,3 — 0,25%.
Миелограмма — в стернальном пунктате определяются характерные признаки гиперплазии красного кроветворного ростка — увеличение количества эритрокариоцитов. Гранулоцитарный и мегакариоцитарный ростки не изменены.
Продолжительность жизни эритроцитов — отмечается значительное сокращение (по данным теста с радиоактивным хромом).
Анализ кала — высокое содержание стеркобилина.
ЭКГ- снижение амплитуды зубца Т в нескольких грудных, нередко стандартных отведениях (вследствие миокардиодистрофии).
УЗИ органов брюшной полости — увеличение селезенки, камни в желчном пузыре. При длительно существующем микросфероцитозе и частых обострениях возможно увеличение печени (вследствие нарушения оттока и застоя желчи).
Рентгенологическое исследование черепа — обнаруживается значительное расширение диплоитического пространства с рисунком в виде «щетки». Признак неспецифический, может наблюдаться и при других видах наследственных гемолитических анемий.
Течение заболевания волнообразное с периодическими обострениями и ремиссиями. Продолжительность ремиссий колеблется от нескольких месяцев до нескольких лет. Обострение заболевания провоцируется психо-эмоциональными стрессовыми ситуациями, тяжелыми физическими нагрузками, вирусными инфекциями и другими факторами. Обострение заболевания характеризуется усилением анемии, желтухи, появлением слабости, иногда болей в животе. При длительном многолетнем течении болезни формируются камни в желчном пузыре. Наиболее тяжелые обострения связаны с гемолитическими кризами (описаны выше).
В некоторых случаях обострение заболевания может проявляться в виде арегенераторных кризов с симптомами гипоплазии преимущественно красного кроветворного ростка. Развитие этих кризов объясняется значительным усилением распада эритроцитов на определенном этапе развития заболевания, резким усилением тормозящего влияния селезенки на кроветворение (вторичный гиперспленизм). В некоторых случаях причиной арегенераторных кризов может быть аллергическая реакция, влияние инфекции, ионизирующей радиации на костный мозг с избирательным угнетением в костном мозге красного кроветворного ростка.
источник
Наследственная микросфероцитарная гемолитическая анемия (болезнь Минковского-Шоффара) — наследственное заболевание, в основе которого лежит дефект белков мембраны эритроцитов — спектрина, анкирина, а также протеинов 4,2 и 3, что приводит к изменению формы (микросфероцитоз), укорочению продолжительности жизни эритроцитов и их разрушению.
Этиология. Заболевание носит обычно семейно-наследственный характер и передается по аутосомно-доминантному типу.
Эпидемиология. Болезнь распространена в средней и северной полосе России с частотой 2,2 случая на 10 000 населения.
Сводится к по вышенному внутриклеточному гемолизу, происходящему в органах ретикулоэндотелиальной системы, главным образом в селезенке и в меньшей степени в печени, костном мозге и лимфатических узлах. Непосредственная причина гемолиза — генетически обусловленная эритроцитопатия, связанная с дефектом белков цитоскелета эритроцитов. Это приводит к нарушению проницаемости для натрия и образованию сферичных эритроцитов, в силу чего продолжительность их жизни значительно укорачивается (до 7-14 дней вместо 120 в норме).
Клинические проявления
Первые симптомы болезни проявляются обычно в детском возрасте. Обращает внимание желтуха при отсутствии других признаков заболевания. В тяжелых случаях, сопровождающихся частыми гемолитическими кризами, рано появляются симптомы анемии: слабость, головокружение, сердцебиение, потеря аппетита.
Гемолитические кризы возникают под влиянием различных провоцирующих моментов (охлаждение, переутомление, травма, беременность, инфекции и др.) и характеризуются ознобами, повышением температуры, усилением желтухи, резким снижением количества эритроцитов и гемоглобина в крови, увеличением размеров селезенки.
При объективном исследовании отмечается лимонно-желтая окраска кожи и слизистых оболочек, обусловленная увеличенным содержанием в сыворотке крови непрямого билирубина. Значительная часть последнего после трансформации в прямой билирубин с желчью поступает в кишечник, последовательно превращаясь в уробилин и стеркобилин, в результате чего испражнения больных окрашиваются в темно-коричневый цвет. Часть уробилина, всасываясь в кишечнике, направляется по воротной вене в печень, но так как последняя в силу перегрузки не в состоянии перевести все количество уробилина в билирубин, то часть уробилина, минуя печень, поступает в кровь, а затем выделяется с мочой, придавая ей цвет пива или крепкого чая. Моча при гемолитической желтухе не содержит билирубина, так как он находится в крови не в свободном состоянии, а в соединении с белком.
Вторым признаком болезни является увеличение селезенки и в меньшей степени печени за счет резкой гиперплазии ретикулоэндотелиальной системы. При пальпации селезенка плотная, безболезненная и может достигать огромных размеров (1-2 кг).
Часто отмечается склонность к образованию камней в желчном пузыре и желчевыводящих путях с развитием клиники ЖКБ. Выпадению камней в желчном пузыре способствует сгущение желчи, богатой пигментами (плейохромия).
Нередко у больных наблюдаются аномалии развития скелета (башенный квадратный череп, седловидный нос, высокое стояние твердого нёба, отставание в росте, укорочение мизинцев, микрофтальмия и др.) и трофические язвы голени. Последние обусловлены нарушением кровоснабжения конечностей вследствие гемолиза эритроцитов и образования тромбов в капиллярах.
Гематологическая характеристика. Важным признаком заболевания является шарообразная форма эритроцитов без просветления в центре —сфероцитоз, а также микронитоз (диаметр эритроцитов менее 6,5 мкм). В легких случаях заболевания общее количество эритроцитов и гемоглобина не снижается, поскольку усиленная деятельность костного мозга обеспечивает компенсацию повышенного распада эритроцитов. При резко выраженном гемолизе, особенно во время кризов, анемия нарастает. Количество ретикулоцитов повышается до 50-100%о, а в отдельных случаях превышает даже 50%.
Существенно снижена осмотическая устойчивость эритроцитов к разведению — до 0,70-0,50 вместо нормы 0,48-0,32.
Течение болезни волнообразное, со сменой светлых промежутков периодами обострений, сопровождающихся гемолитическими кризами.
Прогноз болезни благоприятен. При отсутствии значительной анемии больные сохраняют трудоспособность в течение многих лет. Однако у некоторых лиц развивается калькулезный холецистит, сопровождающийся частыми приступами печеночной колики. В более редких случаях заболевание протекает с частыми гемолитическими кризами, что ведет к развитию анемии, физической и умственной отсталости, снижению трудоспособности больных.
Диагноз врожденной гемолитической анемии болезни (Минковского-Шоффара) ставится на основании характерной триады — гемолитической желтухи, спленомегалии и гиперрегенераторной анемии.
При дифференциальной диагностике следует иметь в виду приобретенную хроническую гемолитическую анемию типа Гайем-Видаля, а также острые гемолитические анемии, протекающие преимущественно с внутрисосудистым гемолизом.
В клинической картине врожденной и приобретенной гемолитических анемий имеется много общих черт: цикличность течения, наличие желтухи, увеличение содержания непрямого билирубина в сыворотке крови, анемия, ретикулоцитоз, эритробластическая реакция костного мозга и т. д. Приобретенная гемолитическая анемия, в отличие от болезни Минковского-Шоффара, характеризуется тяжелым течением и более выраженной анемией — количество эритроцитов снижается до 2х10 12/л , а НЬ — до 50 г/л. По образному выражению Шоффара, эти больные «более бледны, чем желтушны». В анамнезе у них отсутствуют указания на семейный характер заболевания. При этой форме анемии в крови нередко обнаруживаются антиэритроцитарные агглютинины (положительная проба Кумбса). При врожденной гемолитической анемии спленэктомия дает почти стопроцентный терапевтический эффект, а при приобретенной она обеспечивает неполное выздоровление, причем только в 50% случаев.
Дифференциальный диагноз более подробно представлен в таблице.
От острых гемолитических анемий болезнь Минковского-Шоффара отличается семейно-наследственным характером заболевания, цикличностью течения, наличием типичных гематологических признаков и отрицательной пробой Кумбса.
В отдельных случаях врожденную гемолитическую анемию приходится дифференцировать с билиарным циррозом печени. При этом помогают такие признаки, как повышение количества непрямого билирубина, а главное -наличие типичной гематологической триады (понижение осмотической резистентности эритроцитов, микросфероцитоз, высокий ретикулоцитоз). Кроме того, при болезни Минковского-Шоффара обычно отсутствуют симптомы поражения печени и портальной гипертензии.
источник
Основные этапы патогенеза наследственного микросфероцитоза
Изменение белка в мембране эритроцита — первопричина дефекта красных кровяных клеток; нарушение транспорта катионов — вторично. Этой точки зрения в настоящее время придерживается преобладающее число исследователей. Существует мнение, что изменения белка вторичны, поскольку они обнаруживаются не только при наследственном сфероцитозе, но и при аутоиммунной гемолитической анемии. Обобщая данные литературы, основные патогенетические звенья наследственного микросфероцитоза можно представить в следующем виде. Наследственный дефект мембраны эритроцита приводит к повышенной проницаемости ее для ионов натрия, что, в свою очередь, содействует возрастанию интенсивности гликолиза, повышению интенсивности метаболизма липидов, потере поверхностных субстанций, изменению объема клетки, формированию стадии макроцита. Макроциты при движении на уровне селезенки начинают испытывать механическое затруднение, в связи с чем они длительно задерживаются в красной пульпе, подвергаясь всем видам неблагоприятных воздействий (гемоконцентрация, изменение рН, активная фагоцитарная система). Неблагоприятные условия обмена в селезенке способствуют повреждению мембраны, что еще более увеличивает сферичность клетки и содействует формированию стадии микроцитов. Уменьшенный внутриклеточный рН микросфероцитов способствует торможению их гликолитической активности в условиях недостаточного снабжения глюкозой в микрососудах селезенки, что сопровождается снижением активности транспорта ионов, повышением осмотического содержания клетки и осмотическим лизисом. Селезенка при данном заболевании, по мнению ряда авторов, активно наносит эритроцитам повреждение, содействуя еще большей фрагментации эритроцитарной мембраны и сферуляции. Этот факт нашел подтверждение в электронно-микроскопических исследованиях, которые обнаружили в эритроците ультраструктурные изменения, выражающиеся утолщением клеточной мембраны, ее разрывами и образованием вакуолей. Через 2—3 пассажа через селезенку микросфероцит подвергается лизису и фагоцитозу. Фагоцитарная гиперактивность селезенки, в свою очередь, вызывает прогрессирующую гиперплазию органа и дальнейшее повышение его фагоцитарной активности. Нормализация срока жизни эритроцитов после операции свидетельствует о том, что только фагоцитарная активность селезенки опасна для сфероцита, печень же в этом отношении остается интактной. То же самое подтверждается и исследованиями с радиоактивным хромом, выявляющими резкое повышение радиоактивности печени и селезенки при аутоиммунных гемолитических анемиях и только селезенки — при сфероцитозе. Следовательно, при сфероцитозе гемолиз зависит в основном от формы эритроцита. Селезенка является местом деформации и гибели эритроцитов. Гемолитический процесс при наследственном микросфероцитозе приводит к анемии и гипоксии, гиперцеллюлярной реакции костного мозга с выбросом в периферическую кровь эритроидных клеток, усиленному образованию и экскреции желчных пигментов. Большой интерес представляют работы, в которых было показано, что в эритроцитах больных наследственным микросфероцитозом, инкубируемых в среде без глюкозы, происходит прогрессивное снижение содержания липидов (преимущественно холестерола, сфингомиелина й лецитина), которое предшествовало уменьшению осмотической стойкости. Добавление глюкозы замедляло, но не предупреждало потери клеточных липидов сфероцитами. Фосфолипиды, как установлено, принимают участие в транспорте катионов через клеточную мембрану, и обмен их ускоряется при увеличении скорости вхождения натрия в клетку. Эти компоненты необходимы для поддержания постоянства структуры липопротеидного слоя мембраны, и ускоренный их метаболизм в сфероцитах, обусловленный повышенной скоростью транспорта натрия, приводит к потере мембранных компонентов клетки. В эритроцитах, утрачивающих как холестерин, так и фосфолипиды (что провоцируется нарушением гемостаза, в первую очередь в отношении глюкозы, и приводит к прогрессированию микросфероцитоза), нарушения мембраны необратимы, и такие клетки нежизнеспособны in vivo. Определенное значение в изменении формы эритроцитов имеет пониженное содержание в клетке АТФ, так как механические свойства эритроцитов (способность к деформации и фильтруемость) резко уменьшаются при падении уровня этого макроэрга в клетке, что сопровождается появлением микросфероцитоза. Эритроциты больных наследственным микросфероцитозом обладают, таким образом, следующими особенностями метаболизма: повышенным аутогемолизом, частично корригируемым глюкозой и АТФ, увеличенной скоростью гликолиза (последний аномально чувствителен к лишению глюкозы), повышенной скоростью прохождения натрия через клеточную мембрану, увеличенной потерей холестерина при инкубации в среде, содержащей глюкозу, и ускоренной и равномерной утратой липидов (холестерина и фракций фосфолипидов) при инкубации этих клеток в среде, лишенной глюкозы. Разрушение эритроцитов начинается в периферической крови и заканчивается в макрофагах, в которых из гемоглобина образуется билирубин и выделяется ими в периферическую кровь. Этот неконъюгированный (свободный) билирубин не выводится почками, поскольку содержит высокомолекулярное соединение глобин, задерживаемый внутренним слоем капсулы Шумлянского— Боумена. С током крови билирубин попадает в печень, там гепатоциты отщепляют глобин и образуют новое соединение, состоящее из порфириновой цепи. Это соединение выделяется желчью и называется конъюгированным билирубином. Являясь низкомолекулярным соединением, последний свободно проходит почечный фильтр. Неконъюгированный билирубин (дает «непрямую» реакцию с диазореактивом), нерастворимый в воде, в печеночной клетке соединяется с глюкуроновой кислотой, которая придает ему растворимость в воде, способность проходить через почечный фильтр и быструю (прямую) реакцию с диазореактивом. Неконъюгированный билирубин (гемобилирубин) в больших концентрациях токсичен, растворяется в жирах и легко проникает в нервные клетки коры головного мозга, расстраивая в них процессы окислительного фосфорилирования. Для проникновения неконъюгированного билирубина в печеночную клетку необходимо-наличие активного фермента глюкуронилтрансферазы. Таким образом, уровень гипербилирубинемии зависит как от количества внутриклеточно распадающихся эритроцитов, так и от функциональных способностей печеночной клетки «обезвреживать» этот билирубин, переводить его в водорастворимый билирубиндиглюкуронид. КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ. Первые признаки заболевания могут проявиться и в детском возрасте, но чаще — в юношеском и зрелом. При заболевании микросфероцитозом отмечают желтуху, анемию, спленомегалию, изменение скелета. В течение длительного времени единственным признаком заболевания является желтушное окрашивание склер и кожи. Течение волнообразное. Причиной усиления гемолиза и, соответственно, ухудшения состояния чаще всего является инфекция, переохлаждение, беременность. Развивается слабость, появляется одышка и учащенное сердцебиение при физической нагрузке. Степень интенсивности желтухи может быть различной: от незначительной до резки выраженной. С каждым обострением желтушность усиливается. У детей первых месяцев жизни с функциональной слабостью гепатоцитов гипербилирубинемия бывает особенно высокой с резко выраженной желтухой и поражением ядер головного мозга (ядерная желтуха). У детей старшего возраста проявление заболевания (кризов) нередко осложняется желчно-каменной болезнью, причем билирубиновые камни при рентгеноскопическом исследовании не обнаруживаются.
источник
При анемии в крови резко снижается уровень гемоглобина. С этим заболеванием в течение всей жизни приходится сталкиваться многим людям. Более того, не обходит оно стороной даже маленьких детей. Большинство разновидностей недуга обусловлены плохим питанием, дефицитом витаминов или приемом некоторых препаратов. После устранения провоцирующего фактора все симптомы купируются. Среди всего многообразия патологических процессов выделяется один сложный и весьма опасный. Это гемолитическая анемия Минковского-Шоффара. Именно о ней речь пойдет в сегодняшней статье.
Под анемией принято понимать состояние организма, характеризующееся резким снижением показателей эритроцитов и гемоглобина. Некоторые разновидности заболевания приводят к изменению формы красных кровяных клеток. С течением времени они утрачивают первоочередные функции.
Анемия часто сопровождает различные заболевания, но никогда не бывает первичной. Именно поэтому не стоит оставлять расстройство без внимания. Необходимо как можно быстрее найти его причину и постараться устранить.
Понятие «гемолитической анемии» включает в себя обширную группу заболеваний. Все они характеризуются общим патогенезом. Усиленное разрушение эритроцитов приводит к увеличению продуктов их распада и к нарастанию эритропоэза. Цикл формирования красных телец нарушается. Процессы их разрушения постепенно начинают превалировать над механизмами появления и вызревания.
Все гемолитические анемии условно подразделяются на две группы: наследственные и приобретенные. В этой статье мы более подробно остановимся на первом варианте. Если быть точнее, то рассмотрим, что представляет собой наследственная анемия Минковского-Шоффара.
В медицинских справочниках можно встретить несколько названий, описывающих патологический процесс. Это и микросфероцитарная анемия, и наследственный сфероцитоз, и болезнь Минковского-Шоффара. Чаще всего используется последнее наименование по фамилиям ученых-первооткрывателей.
Данное заболевание считается весьма распространенным (1 случай на каждые 5 тыс. населения). Диагностируется преимущественно у жителей Северной Европы. Первые признаки становятся заметны у детей в раннем возрасте. Если не приступить к лечению недуга своевременно, его течение негативно отразится на функционировании всего организма.
Анемия Минковского-Шоффара сопровождается нарушением структуры и функций клеточной мембраны эритроцитов. В результате происходящих процессов они меняют свою форму на круглую, становятся хрупкими. Появляются первые признаки гемолиза — разрушения красных кровяных клеток с одновременным выделением гемоглобина.
У здорового человека эритроциты по своей форме напоминают двояковогнутый диск, благодаря чему они беспрепятственно передвигаются по сосудам. При анемии в мембране этих элементов синтез белка нарушается. Это приводит к проникновению жидкости внутрь клеток. По этой причине они меняют свою форму. Проходя через сосуды, эритроциты сильно деформируются, а через некоторое время начинают разрушаться. На фоне происходящих процессов уровень красных кровяных элементов резко падает, развивается гемолитическая анемия.
Если у одного из родителей ранее уже был диагностирован этот недуг, он обязательно перейдет по наследству ребенку. Крайне редко больные дети рождаются у совершенно здоровых мам и пап. В этом случае анемия развивается на фоне изменений в ДНК. Первичная генная мутация происходит еще во время внутриутробного развития плода.
Обязательным условием развития болезни является воздействие на организм матери следующих факторов:
- радиация, рентгеновское излучение;
- интоксикация солями тяжелых металлов, наркотическими веществами, никотином;
- вирусная атака.

Клиническая картина во многом определяется степенью тяжести патологического процесса и количеством измененных эритроцитов. Первые его симптомы можно наблюдать у детей дошкольного и раннего школьного возраста. Течение анемии этого вида обычно волнообразное. Приступы гемолитического криза, когда происходит одновременное разрушение большого числа эритроцитов, сменяются периодами затишья. При этом симптомы могут чуть отличаться.
Например, межприступный период болезни проявляется признаками анемии. Среди них можно выделить бледность кожных покровов, слизистых и склер глаз. При гемолитическом кризе клиническая картина видоизменяется и сопровождается следующими симптомами:
- Повышение температуры до 38 градусов, головная боль, общее недомогание.
- Развитие желтухи.
- Боль в животе, отличающаяся спастическим характером.
- Дискомфорт в области печени из-за ее увеличения.
- Воспаление селезенки.
Наследственная гемолитическая анемия Минковского-Шоффара встречается и у взрослых. Наиболее частой причиной обращения к врачу в этом случае служит желтушность кожных покровов. Однако в большинстве случаев этот недуг протекает бессимптомно. Пациенты узнают о его существовании случайно и обычно во время профилактического осмотра.
Диагностика анемии Минковского-Шоффара достаточно проста. При подозрении на заболевание и появлении начальных его признаков следует обратиться за помощью к врачу. Патологии кроветворной системы находятся в компетенции гематолога. После изучения жалоб пациента и его семейного анамнеза специалист должен осмотреть кожные покровы и склеры, провести пальпацию живота. В обязательном порядке назначается УЗИ печени и селезенки, поскольку одним из симптомов недуга является увеличение в размерах этих органов.
Одновременно гематолог дает направление на ряд лабораторных анализов. Гемолитическая анемия Минковского-Шоффара подтверждается при наличии следующих изменений:
- Моча: гемоглобинурия, повышение показателей белка и уробилина.
- Биохимия крови: снижение холестерина, рост лактатдегидрогеназы, увеличение непрямого билирубина.
- Исследование эритроцитов: выраженный ретикулоцитоз, сокращение размеров клеток, понижение их осмотической устойчивости.
- Общий анализ крови: ускорение СОЭ, незначительное снижение тромбоцитов и лейкоцитов, сокращение цветового показателя.
Гемолитическая анемия Минковского-Шоффара у детей иногда вызывает трудности во время диагностики. Это заболевание имеет схожие с другими аутоиммунными патологиями симптомы. Поэтому врачи должны знать некоторые отличительные и характерные именно для данного вида анемии признаки.
В первую очередь речь идет о наследственной предрасположенности. Только в исключительных случаях оба родителя оказываются абсолютно здоровыми. С другой стороны, у больного ребенка прослеживаются явные изменения в костях черепа. В сомнительных случаях дополнительно назначается проба Кумбса. Если анализ отрицательный, у пациента подтверждается анемия Минковского-Шоффара. Диагностика на этом считается завершенной.
Терапия анемии подбирается с учетом ее тяжести. В период затишья, как правило, вмешательство не требуется. Во время очередного приступа больного сразу госпитализируют.
Консервативное лечение анемии Минковского-Шоффара включает в себя следующие меры воздействия:
- Заместительная терапия эритроцитарной массой, если уровень гемоглобина в крови падает до отметки 70 г/л.
- Лечение альбуминами назначается при высоких показателях билирубина.
- Для дезинтоксикации организма используется инфузионная терапия.
- В период отсутствия выраженного гемолитического криза показан прием желчегонных препаратов.
Продолжительность такой терапии, конкретные препараты и их дозировка — все эти вопросы решает врач в индивидуальном порядке.
Если микросфероцитарная гемолитическая анемия Минковского-Шоффара протекает в тяжелой форме, консервативное лечение не справляется с заявленными задачами, пациенту рекомендуется операция по удалению селезенки. Такой подход не позволяет полностью вылечить недуг. С другой стороны, после вмешательства заметно сокращается число разрушенных эритроцитов, а их жизненный цикл удлиняется.
Гемолитические кризы после операции не повторяются, но она имеет ряд противопоказаний. Например, удаление селезенки не рекомендуется детям младше 5 лет по причине высокого уровня смертности в послеоперационный период. Негативной стороной процедуры считается снижение сопротивляемости организма по отношению к вирусным и грибковым инфекциям.
Альтернативным вариантом удаления селезенки считается эндоваскулярная окклюзия. Это еще один метод лечения, к помощи которого часто прибегают при диагнозе «микросфероцитарная анемия Минковского-Шоффара». В ходе процедуры врач вводит в орган лекарство, которое провоцирует спазм и приводит к инфаркту селезенки. Некоторая ее часть после этого сохраняет полноценное кровоснабжение и не утрачивает способность сопротивляться инфекциям.
Гемолитическая анемия Минковского-Шоффара у детей дошкольного возраста часто приводит к отставанию в психическом и физическом развитии. Особенно если родители долго не обращались за медицинской помощью или игнорировали рекомендации врача.
У взрослых пациентов наиболее распространенным осложнением считается желчнокаменная болезнь на фоне нарушения билирубинового обмена. Все дело в том, что гемолитический криз часто воспринимают за начало развития механической желтухи, поэтому правильное лечение откладывается. При наличии камней в желчном пузыре рекомендуется проведение холецистэктомии вместе со спленэктомией.
При легком течении заболевания и своевременно проведенной операции по удалению селезенки прогноз благоприятный. Ремиссия обычно наступает сразу после гемолитического криза. Ее продолжительность может варьироваться, но в большинстве случаев составляет около двух лет.
Анемия Минковского-Шоффара носит наследственный характер. Поэтому предупредить возникновение заболевания не представляется возможным. В целях профилактики тяжелых форм недуга пациентам с выявленной формой анемии рекомендуется периодически проходить полное обследование у гематолога.
При планировании беременности следует понимать, что вероятность развития заболевания у будущего ребенка составляет 50%. Поэтому новорожденному также показано постоянное наблюдение у врача для выявления патологии на ранней стадии.
источник
Врожденная микросфероцитарная анемия — гемолитическая анемия, в которой нарушается структура эритроцитов, то есть красных кровяных телец. Тахикардия и спленомегалия являются типичными симптомами наследственного гемолитического гепатита. У ребенка они обнаруживаются сразу после рождения. Также проводится диагностика заболевания в основном на результатах анализа крови с мазком, мочой. Болезнь нельзя вылечить, терапия уменьшает симптомы.
Микросфероцитарная анемия относится к группе гемолитической анемии. В классификации ICD-10 она упоминается как врожденная гемолитическая желтуха без выделения желчных мочевых путей, а также анемия Минковского-Шоффара. Она может быть унаследована, причем врожденная гемолитическая анемия диагностируется гораздо чаще.
Это наиболее распространенный вид наследственных анемий, возникающих с частотой 1 на 5000 человек. Из-за генетической мутации существует дефицит белков, которые создают мембраны эритроцитов — они принимают сферическую форму. Это приводит к их обширному разрушению в селезенке и в просвете кровеносных сосудов.

В 10-20% болезнь не наследуются, и мутация появляется спонтанно. Предварительно приобретенная анемия Минковского-Шоффара у детей может сопровождаться аутоиммунной гемолитической анемией, когда антитела разрушают эритроциты, неправильно признавая их чужеродными. Поэтому нельзя предотвратить развитие микросфероцитарной анемии и провести эффективную профилактику. Вот почему так важно диагностировать анемию Минковского-Шоффара как можно скорее, а затем осуществить соответствующее лечение.
Симптомы сфероцитарной анемии могут иметь разную степень тяжести, что является основой для классификации болезни при тяжелом, умеренном или легком течении. Этот тип гемолитической анемии может характеризоваться едва заметными симптомами, особенно в легкой форме заболевания. Если в семье присутствует врожденная микросфероцитарная анемия, симптомы не должны усиливаться.
Наиболее распространенными симптомами анемии Минковского-Шоффара являются:
- пожелтение кожи и слизистых оболочек (гемолитическая желтуха);
- увеличение селезенки (спленомегалия);
- холелитиаз как осложнение хронического гемолиза;
- общие симптомы гемолитической анемии: слабость, расстройства концентрации, головные боли и головокружение, плохая переносимость, ускоренный пульс.
Симптомы врожденной микросфероцитарной гемолитической анемии у детей появляются сразу после рождения или в раннем детстве. В крайних случаях болезнь приводит к смерти.
В дополнение к возникновению характерных симптомов и положительной семейной истории результаты лабораторных тестов имеют ключевое значение.
Исследования по наследственной микросфероцитарной анемии включают:
- Анализ крови с мазком, где можно заметить изменения, характерные для анемии: пониженные эритроциты (низкий уровень эритроцитов), сниженный гемоглобин; в мазке видны сфероциты, то есть сферические эритроциты и фрагменты дезинтегрированных клеток крови и повышенный ретикулоцитил.
- Биохимический анализ крови на увеличение билирубина, повышенную лактатдегидрогеназу, увеличение калия (гиперкалиемия) и гаптоглобина в крови.
- Анализ мочи, указывающий на присутствие уробилиногена, и определение стула стериколиногена.

Характерным исследованием, подтверждающим наследственную гемолитическую анемию, является тест на резистентность эритроцитов и тест с подкисленным глицерином. Эти тесты включают лечение крови пациента гипотоническим хлоридом натрия и глицерином, определение степени гемолиза эритроцитов.
Чтобы проверить, врожденная или приобретенная гемолитическая анемия, проводится прямой тест на антиглобулины Кумба, что дает положительный результат только в случае приобретенной анемии Минковского-Шоффара.
Кроме того, проводятся визуализационные обследования, такие как компьютерная томография или ультразвук брюшной полости и рентгенография грудной клетки.
В случае наследственной микросфероцитарной гемолитической анемии лечение невозможно, как и устранение ее причин. Если симптомы наследственной гемолитической желтухи усиливаются, используется лечение, типичное для каждой анемии: переливание концентрата эритроцитов, стероидная терапия, инъекция эритропоэтина, стимулирующего синтез эритроцитов.
Спленэктомия предназначена только для самых тяжелых случаев, особенно при холелитиазе (тогда фолликул также удаляется). Удаление селезенки не проводится у ребенка в возрасте до 7-8 лет.
Для профилактики послеоперационной инфекции все пациенты, получившие разрешение на спленэктомию, вакцинируются против пневмококков, менингококков и гемофилов.
источник
Наследственный микросфероцитоз (болезнь Минковского-Шоффара)
Это анемия, связанная с генетическим дефектом мембраны эритроцитов, что обусловливает их микросфероцитарную форму и гемолиз.
Тип наследования — аутосомно-доминантный.
Нарушение структурного белка (спектрина) ведет к изменению клеточной мембраны, изменению формы эритроцитов.
При данном заболевании наблюдается повышение проницаемости эритроцита для натрия, что приводит к проникновению избытка натрия и воды в клетку. Эритроциты приобретают сферическую форму, что нарушает их способность деформироваться в узких участках кровотока, например, при переходе из межсинусных пространств селезенки в синусы. Здесь эритроциты повреждаются и разрушаются макрофагами селезенки. Таким образом, тип гемолиза при наследственном микросфероцитозе — внутриклеточный. В крови нарастает анемия и билирубинемия. Длительно существующий гемолиз ведет к стимуляции эритропоэза, у больных нарушается костеобразование: «башенный череп», «готическое небо», монголоидный тип лица.
Клиника
1. Первые клинические симптомы анемии чаще проявляются в детском возрасте.
2. Желтуха, увеличение селезенки, боли в левом подреберье, иногда увеличение печени, склонность к образованию камней в желчном пузыре (желчная колика), изредка симметричные язвы голеней.
3. Деформации скелета («башенный череп», «готическое небо», деформация челюстей с неправильным расположением зубов, полидактилия), микрофтальмия.
4. Периодически возникают гемолитические кризы (часто провоцируются инфекцией): боли в области печени, селезенки и их увеличение, озноб, потемнение мочи, повышение температуры тела до 39—40 ºС, рвота, усиление желтухи и анемии, лейкоцитоз со сдвигом влево, в крови появляются нормоциты или нормобласты.
5. Иногда могут возникать апластические кризы, вызванные инфицированием парвовирусом (PV) — быстро нарастает арегенераторная анемия, иногда ведущая к летальному исходу. Если же происходит выживание, возникает пожизненный иммунитет, обеспечиваемый противовирусным IgG.
Лабораторные данные
1. В крови — нормохромная, микросфероцитарная, гиперрегенераторная анемия, возможны признаки гиперспленизма (тромбоцитопения), например: эр. — 2,1 • 10 12 /л, Hb — 68 г/л, ЦП — 1,0, ретик. — 56 ‰, тромб. — 190 • 10 9 /л, лейк. — 6,1 • 10 9 /л: э — 1, п — 4, с — 60, л — 30, м — 5, СОЭ — 12 мм/ч, анизоцитоз +++, Ht — 30 %, микросфероциты.
2. Снижение осмотической резистентности эритроцитов (в норме минимальный гемолиз 0,48—0,46 % NaCl, максимальный гемолиз — 0,34—0,32 % NaCl). Например, min — 0,64 %, max — 0,42 % NaCl.
3. Костный мозг: раздражение красного ростка (преобладание эритробластов и нормоцитов).
4. В крови — повышение билирубина за счет свободного, увеличение активности ЛДГ.
5. В моче много уробилина, в кале — стеркобилина (плейохромия).
6. Специальный метод исследования: определение продолжительности жизни эритроцитов с помощью радиоактивного хрома (время полувыведения 5—6 дней при норме 22—30).
7. Рентгенологически: изменение костей («волосатый череп»).
Дифференциальная диагностика проводится с АИГА, при апластическом кризе с АА.
1. Наиболее эффективным методом лечения является спленэктомия (при наличии конкрементов в ЖВП — их удаление), приводящая к клиническому выздоровлению.
Показания к спленэктомии (рекомендуется проводить в возрасте 7—8 лет и старше):
— выраженная анемия с частыми гемолитическими кризами, гиперспленизмом и спленомегалией (может быть показанием к операции в любом возрасте);
— вторичная желчнокаменная болезнь (при ее наличии одновременно проводится холецистоэктомия);
— упорная желтуха.
2. Переливание эритроцитарной массы производится только по жизненным показаниям — при крайне тяжелой степени анемии, в период тяжелых гемолитических кризов, а также при выраженной анемии и отказе от спленэктомии. Фолиевая кислота курсами.
3. При апластическом кризе Ig человека для в/в введения в дозе 1—2 г/сут. в течение 5 дней.
4. Дезагреганты и антикоагулянты для профилактики тромбозов (Флебодиа).
источник