Многие люди знают, что анемия является довольно опасным заболеванием. Существует множество разновидностей этого недуга, и каждый из них приносит вред здоровью человека. Сидеробластная анемия представляет собой опасную патологию, связанную с нарушением синтеза микроэлементов. При данном заболевании костный мозг использует железо для объединения гемоглобина, поэтому оно откладывается во внутренних органах. Главное – не допустить осложнений, а для этого необходимо своевременно обратиться за помощью к специалисту.
Сидеробластная анемия отличается от других видов заболевания сниженной концентрацией железа в эритроцитах. Дело в том, что костный мозг не задействует этот элемент в синтезе гемоглобина. Чаще всего недуг развивается как врождённый или приобретённый. На генетическом уровне анемия встречается в основном у мальчиков.
Заболевание может передаваться также аутосомно-доминантным путём. Эту анемию называют синдромом Пирсона. Когда в организме плохо усваивается железо, оно откладывается во внутренних органах, что приводит к сидеробластной анемии. Если железа становится слишком много, нарушается работа печени, почек и сердечной мышцы.
Анемия этого типа подразделяется по степени тяжести, а также по причине появления и клинической картине. Выделяют несколько видов сидеробластной анемии:
- Наследственная. Недуг переходит по наследству в результате мутации гена. Эту патологию вызывает аномалия процесса обмена веществ при участии витамина В6 и аминолевулиновой кислоты. Заболевание даёт о себе знать после рождения или в подростковом возрасте.
- Врождённая. Эту форму выделяют отдельно, хотя в какой-то степени она относится к наследственному виду. Здесь характерно высокое содержание эритроцитарного копропорфирина.
- Приобретённая. Появляется в результате неблагоприятного воздействия химических веществ. Среди них выделяют этанол, свинец, циклосирин.
Проблемы с синтезом железа возникают также по причине опухолевых процессов в организме. Примерно 1/10 часть больных сидеробластной анемией страдает острым лейкозом.
Основной причиной развития этого заболевания является нехватка протопорфиирина, который является одним из компонентов синтеза наиболее важного элемента гемоглобина. Кроме этого вещества, в создании участвует ещё белок и железо.
Приобретённая форма сидеробластной анемии возникает из-за того, что в организм не поступают необходимые вещества в достаточном количестве. Бывают ситуации, когда нужные соединения подавляются медикаментами, которые принимает человек.
Организм истощается в результате воздействия алкоголя. Анемия может появиться из-за свинцового отравления или приёма сильных антибиотиков. Наследственная форма передаётся через женскую хромосому с повреждённым геном. Также причиной возникновения заболевания может служить расстройство иммунной системы и развитие опухолевых процессов.
Процесс выявления этого заболевания довольно сложный, так как оно протекает практически бессимптомно. Более того, нет ярко выраженной клинической картины, на которую можно опираться. Если смотреть только на внешние признаки, то диагностировать эту болезнь нереально. Однако есть один способ обнаружения сидеробластной анемии – анализ крови.
Также практикуют обследование внутренних органов пациента, чтобы найти отложения железа. Но к этому времени элемент и так вызывает гематологические симптомы. Чтобы не ошибиться, необходимо подтвердить диагноз. Это делается с помощью макроскопического исследования костного мозга.
Для того чтобы риск сводился к минимуму, необходимо провести биопсию. Именно эта процедура является самым эффективным способом определения сидеробластной анемии. Она осуществляется следующим образом: биоптат предварительно окрашивают специальным веществом, и, если обнаруживается несинтезированное железо, становятся видны характерные соединения.
Как уже было отмечено, симптомы сидеробластной анемии практически не определяются. Клиническая картина также отсутствует, и из-за этого возникают определённые трудности. Если человек чувствует быструю утомляемость, слабость в организме, то он, как правило, обращается к терапевту. Это врач при подозрении на анемию перенаправляет пациента к гематологу, который проводит обследование.
Сначала специалист выясняет общее состояние пациента, интересуется образом жизни, наличием вредных привычек, списком перенесённых заболеваний и т. д. Если врач посчитает нужным, он проведёт несколько исследований. К ним относятся:
- анализ крови (общий и биохимический);
- биопсия печени;
- анализ клеточного состава костного мозга.
Гематолог при необходимости может направить пациента к другим специалистам для более полного обследования. Например, чтобы выяснить форму анемии, нужно обратиться к генетику. Данный врач определит, имеет ли место быть наследственный вид сидеробластной анемии. Также может понадобиться консультация гинеколога, уролога или проктолога.
Прежде чем приступить к терапии, необходимо определить, действительно ли происходит отложение железа. Для этого используют десфераловую пробу. Её вводят внутримышечно, и в результате с мочой должно выделяться около 0,5-1,1 мг железа, а при гипохромной, гиперсидеремической, сидероахрестической, сидеробластной анемии – 5-10 мг.
Стоит отметить, что наследственную форму заболевания вылечить невозможно. Чтобы подавить ген, который начал мутировать, часто используют лечение высокими дозами витамина В6. Его вводят в количестве 100 мг в день. Однако реакция организма здесь практически непредсказуема. В процессе лечения сидеробластной анемии уровень гемоглобина должен повыситься до нормальных показателей за три месяца. Если в этот период времени улучшений не произошло, дальнейшая терапия бессмысленна.
Довольно часто заболевание возникает в результате отравления свинцом. Чтобы это предотвратить, необходимо осторожно обращаться с этим веществом. При реконструкции старых домов нужно соблюдать меры предосторожности, по возможности переселяя временно детей. Запрещается сжигать или закапывать краски, содержащие свинец. Лучше всего соскоблить или удалить их химическим путем. Кроме того, нужно регулярно следить за чистотой жилых помещений, соблюдать строительные и санитарные нормы.
Вылечить наследственную форму заболевания невозможно. Для обеспечения благоприятных последствий необходимо постоянно следить за показателями крови, особенно уровнем гемоглобина. Это достигается путём регулярного прохождения курса терапии, который не позволяет болезни развиваться и поддерживает нормальное состояние человека.
источник
Сидеробластная анемия (врожденная или приобретенная) — это анемия, когда в крови наблюдается избыток железа. Причины включают злоупотребление алкоголем, хронический миелоидный лейкоз или миелодиспластический синдром. Симптомы сидеробластической анемии включают, например, бледную кожу, головные боли и головокружение, быстрое сердцебиение. Лечение в этом случае зависит от типа анемии.
При железодефицитной анемии появляется нехватка железа в организме. Парадоксально, но иногда этот элемент бывает в изобилии, но не может быть использован для синтеза гема.
В этой ситуации развивается сидеропенический синдром, симптомы которого часто напоминают симптомы железодефицитной анемии.
Сидеробластическая анемия является редким заболеванием. Существует врожденная и приобретенная форма.
Сидеропеническая анемия связана с образованием сидеробластов или эритробластов с железным кольцом вокруг ядра. Эритробласты являются предшественниками эритроцитов, которые обычно используют железо для синтеза гема.
Однако если этот процесс нарушен, железо, накопленное в эритробластах, не используется и осаждается в избытке в печени и селезенке. Аномальные непигментированные эритроциты (которые не содержат достаточного количества гемоглобина) циркулируют в кровотоке без достаточного переноса кислорода.
Причинами сидеробластической анемии могут быть генетические дефекты. Такая наследственная анемия выявляется вскоре после рождения, хотя есть случаи, диагностированные только у взрослых.
Приобретенная сидеробластическая анемия может иметь много причин, наиболее распространенными из которых являются:
- лекарства: Хлорамфеникол, Изониазид, Циклосерин;
- отравление свинцом или цинком;
- миелодиспластический синдром;
- злоупотребление алкоголем;
- хронический миелоидный лейкоз.
Симптомы возникают из-за двух основных нарушений: первое — появление небольших непигментированных эритроцитов в крови, которые не переносят достаточное количество кислорода. Второе — накопление железа в органах и мягких тканях, то есть вторичный гемохроматоз.
Симптомы, связанные с тканевой гипоксией, включают головокружение и головные боли, слабость, проблемы с концентрацией. Отмечается бледность кожи и слизистых оболочек, ощущение постоянного холода, ускоренного сердечного ритма и более быстрого дыхания. Накопление железа в печени, селезенке и других органах приводит к диабету, аритмии и неврологическим расстройствам.
Наиболее важную роль играют морфология с мазком, биопсия костного мозга, определение уровня железа в сыворотке, концентрация ферритина и насыщение трансферрина.
Ниже приведены изменения в результатах лабораторных исследований, характерных для сидеробластической анемии:
- в морфологии обнаружено уменьшение количества эритроцитов, уменьшение размера MCV эритроцитов и сниженный гемоглобин в крови;
- в мазке крови присутствуют сидероциты, то есть эритроциты с экстрагемоглобиновыми железными зернами;
- повышенное железо в крови, увеличение ферритина и увеличение насыщения трансферрина.
Результаты анализов крови также указывают на присутствие сидеробластов в костном мозге.

Лечение сидеробластной анемии зависит от ее формы. При врожденной форме используется добавка витамина B6, то же самое относится к сосуществующему недостатку фолиевой кислоты.
Наследственная форма хорошо реагирует на такую терапию — ответ на лечение обычно происходит через несколько недель.
Приобретенная анемия лечится соединениями, которые хелатируют избыток железа в крови (например, дефероксамин).
При лечении очень важно вести правильный образ жизни.
Диета должна исключать продукты, содержащие цинк, что усиливает симптомы заболевания. Поэтому следует избегать чрезмерного потребления печени, ракообразных, орехов, твердых сыров.
Важно избегать употребления алкоголя. Невозможно вылечить врожденную сидеробластную анемию, но при правильном лечении и диете можно жить с ней, не испытывая назойливых симптомов. Однако приобретенные формы сидеробластической анемии полностью излечимы.
источник
Одной из разновидностей гематологических заболеваний является сидеробластная анемия, возникающая на фоне нарушенной переработки внутриклеточного железа, необходимого для синтеза гемоглобина. Как правило, заболевание имеет несколько механизмов развития, и может носить генетический или приобретенный характер. Клиническая картина характеризуется различной степенью тяжести, поэтому чтобы не допустить серьезных осложнений, необходимо провести все терапевтические мероприятия на начальных этапах патологии.
Сидеробластная анемия – это патологическое состояние, также известное, как сидероахрестическая анемия. Оно проявляется дефицитом железа в эритроцитах, вследствие нарушения синтеза микроэлемента костным мозгом.
Для заболевания характерно преобладание микроцитов в периферической крови, а также значительное снижение цветного показателя, что является ее отличительной чертой от железодефицитной анемии. При сидеробластной анемии, железо присутствует в необходимой концентрации, но происходит нарушение механизма попадания микроэлемента в гемоглобин. В дальнейшем вместо железа в клетках, накапливаются сидеробласты, образуя соединения в виде колец.
Главной причиной состояния является дефицит протопорфина, который является важным элементом в процессе синтеза гемоглобина.
Наследственная форма возникает вследствие генетических аномалий, когда происходят патологические изменения в гене или хромосоме, ответственных за развитие заболевания. Также пусковым механизмом в развитии состояния может быть врожденный дефект транспортировки меди, которая необходима для полноценного усвоения железа клетками. В данной ситуации, патология будет носить вторичный характер, поскольку ее сопровождает нарушение обменных процессов в организме.
Как правило, причинной приобретенной формы являются:
- прием медикаментов;
- избыточное употребление спиртосодержащих напитков;
- действие химических веществ;
- отравление тяжелыми металлами;
- гематологические заболевания;
- воспалительные процессы;
- онкологические болезни.
Медикаментозная или токсическая сидеробластная анемия развивается на фоне негативного действия активного компонента лекарства или токсического соединения. Так, при отравлении свинцом заболевание развивается молниеносно, поскольку пары тяжелого металла быстро проникают в ткани организма. При условии незначительного железодефицитного состояния, клиническая картина заболевания протекает в ускоренном темпе.
Способствующие факторы, провоцирующие развитие патологии:
- алкогольная интоксикация;
- злоупотребление лекарственными препаратами;
- аутоиммунные заболевания;
- процесс формирования новообразований;
- аллергия на туберкулостатические лекарства;
- пожилой возраст.
У лиц, страдающих хроническим алкоголизмом, сидеробластная анемия наблюдается в несколько раз чаще, поскольку этанол способствует снижению концентрации фосфатов витамина В6.
Сидеробластическую анемию классифицируют в зависимости от причины возникновении, механизма развития и степени выраженности клинических симптомов.
Различают следующие виды патологического состояния:
- Наследственная . Заболевание передается по женской хромосоме, в результате аномальных изменений в гене. Вследствие дефекта происходит замедление синтеза органических соединений, как гем, приводящего к низкой продукции гемоглобина.
- Врожденная . Аномальные клетки передаются от матери к ребенку. Поэтому женщина с умеренными симптомами заболевания может родить одного здорового ребенка и одного с тяжелыми проявлениями патологии.
- Приобретенная . Развивается на фоне интоксикации лекарственными препаратами и алкоголем. Как правило, после ликвидации раздражителя такая форма анемии устраняется без помощи медикаментов.
Симптоматика заболевания характерна для всех видов и форм анемий:
- Анемический синдром (бледность кожных покровов, сухость слизистых, ломкость ногтевых пластин, головокружение, снижение памяти).
- Нарушения в работе сердца и сосудов (гипотензия, тахикардия, появление систолического шума над верхушкой сердца).
- Расстройство пищеварения (снижение аппетита, искажения вкусовых свойств, нарушения глотания, частые запоры и диарея).
При врачебном осмотре отмечается увеличение печени и селезенки, что обуславливается отложением избыточного количества железа (гемосидероз). Постепенное скопление микроэлемента в печени приводит к развитию цирроза, поэтому на последней стадии заболевания орган уменьшается в размерах, а при ультразвуковой диагностике выявляется уплотнение тканей.
В большинстве случаев, гемосидероз поражает другие системы организма. Так, при накоплении железа в половых органах у мужчин, происходит снижение количества вырабатываемого тестостерона, что приводит к развитию евнухоидизма. При поражении поджелудочной железы развивается сахарный диабет, который может осложниться диабетической комой.
Сидеробластические анемии, вызванные интоксикацией тяжелыми металлами, в клинической картине проявляются острыми болями в брюшной полости, и поражением периферической нервной системы. Очень часто данных пациентов направляют в хирургические отделения с подозрением на острый воспалительный процесс органов брюшной полости.
Диагностика данного вида анемии затруднительна, поскольку протекает без характерных клинических симптомов. Для подтверждения диагноза собирают анамнез жизни пациента, его жалобы, проводят оценку общего состояния, а также назначают прохождение лабораторных и инструментальных методов исследований.
Для диагностики интоксикации свинцом проводиться определение концентрации свинца в венозной крови. В случае хронического отравления тяжелым металлом, проводят рентгенологическое исследование коленных суставов, где обнаруживается в дистальном отделе бедренной кости уплотненные и расширенные участки кальцификации, а в проксимальных отделах выявляют свинцовую линию.
Алгоритм исследование пациента:
- Общий клинический анализ крови (определение количества ретикулоцитов, и характеристикой строения эритроцитов).
- Миелограмма (выявление колец сидеробластов методом окрашивания мазков).
- Биохимический анализ (определение ферментов, почечные и печёночные пробы, глюкоза, сывороточное железо).
- Общий анализ мочи.
- Копрограмма.
- Электрокардиограмма.
- УЗИ органов брюшной полости, сердца, почек
- Рентген коленных суставов.
Для определения формы патологии проводят следующие исследования:
- Дисфераловая проба.
- Концентрация в цельной крови протопорфина эритроцитов.
- Концентрация в цельной крови свинца.
- Проба с ЭДТА.
- Биопсия косного мозга.
Наследственная форма заболевания может иметь различную степень тяжести. С возрастом патология усиливается, приобретая гипохромный характер, когда цветовой показатель снижается до 0,5. В мазке выявляют гипохромные эритроциты, определяется анизоцитоз, пойкилоцитоз.
Наследственная форма недуга не поддается лечению, ее корректируют при помощи симптоматической терапии, направленной на подавление аномального гена. В лечении заболевания противопоказан прием препаратов железа и гемотрансфузии, так как они способны вызвать усугубление гемосидероза внутренних органов.
В качестве основной терапии применяют лекарственное средство пиридоксин, при условии отсутствия резистентности к нему. Дозировка препарата зависит от способа введения, так при внутримышечных инъекциях – 100 мг, внутрь – от 50 до 200 мг два раза в сутки. Терапевтический курс составляет 2 месяца.
Пациентам назначают применять коферменты витамина В6 в высоких дозах, с целью коррекции нарушенной трансформации пиридоксина в пиридоксальфосфат.
При анемии вследствие интоксикации свинцом используют препараты натриевой соли, которые помогают ускорить процесс выведения металла с организма.
В тяжелых случаях, когда отсутствует положительная динамика на медикаментозную терапию, показана заместительная гемотрансфузия красных кровяных тел. Она позволяет добиться временного восстановления баланса гемоглобина и снизить риск развития осложнений. Для предотвращения отложения гемосидерина во внутренних органах, у пациентов, находящихся на гемотрансфузионной терапии, показано использование медикамента Десферала. Его вводят внутривенно в дозе 500–1000 мг, регулярно контролируя уровень железа. Лечение медикаментом рекомендовано проводить небольшими курсами, чтобы не допустить резкого снижения уровня микроэлемента в крови.
Сидеробластические анемии довольно редкая патология. Клиническая картина не имеет отличительных симптомов от других гематологических заболеваний, поэтому необходимо проводить тщательное исследование пациентов. Самым эффективным диагностическим методом в выявлении данного типа анемии является анализ крови, который указывает на развитие патологического процесса в организме. При своевременном лечении можно добиться быстрой стабилизации состояния, однако при врожденной форме прогноз остается неблагоприятным.
источник
Сидероахрестическая анемия (САА) — железонасыщенная или сидеробластная анемия, при которой эритроциты содержат мало железа (гипохромны) вследствие неиспользования его костным мозгом для синтеза гемоглобина.
Этиология и патогенез.В основе развития сидероахрестических анемий лежит нарушение синтеза гема. Железо, белок, необходимые для синтеза гемоглобина, имеются, однако отсутствует достаточное количество протопорфирина. Вследствие этого не осуществляется синтез гема — основного компонента молекулы гемоглобина. Гем — соединение порфири-новых колец (протопорфирина) с атомом железа. Гем, соединяясь с глобином, образует молекулу гемоглобина.
При САА уменьшается образование порфиринов и возникает избыток железа. Уменьшение образования порфиринов обусловлено врожденным или приобретенным дефицитом ряда ферментов. Накопление железа в организме приводит к отложению его во внутренних органах.
Выделяют две основные наследственные формы САА: пиридоксинза-висимую (имеется дефицит пиридоксаль-фосфата, поэтому назначение пи-ридоксина — витамина Be — дает эффект) и пиридоксинрезистентную (этачформа встречается крайне редко).
Приобретенные формы чаще наблюдаются в пожилом возрасте, заболевание не носит семейного характера. Непосредственный ферментный дефект не всегда ясен. САА чаще возникает при лечении туберкулостатичес-кими препаратами вследствие истощения запасов пиридоксаль-фосфата, при свинцовой интоксикации. Могут быть также идиопатические формы САА.
Клиническая картина.При наследственных формах заболевание начинается уже в раннем детстве. На I этапе диагностического поиска выявляются жалобы, обусловленные гипоксически-циркуляторным синдромом. В анамнезе — указания на бледность, слабость, увеличение печени и селезенки. Дети быстро устают, плохо учатся, у них плохая память; у взрослых — слабость, снижение толерантности к физической нагрузке, возникающие после длительного лечения основного заболевания (туберкулеза), профессиональных вредностей (контакт со свинцом). Можно обна-
ружить сведения о выявлении низких показателей гемоглобина и неуспешном лечении препаратами железа.
На II этапе диагностического поиска в периоды обострения возможно выявление бледности кожных покровов и видимых слизистых оболочек, у части больных — увеличение печени и селезенки в умеренных пределах. В связи с этим у таких больных предполагают хроническое заболевание печени (чаще всего хронический гепатит).
Отложение железа во внутренних органах может привести к ряду своеобразных симптомов. Так, отложение железа в поджелудочной железе ведет к сахарному диабету, в печени — к циррозу печени, в сердце — к сердечной недостаточности, в половых железах — к евнухоидизму.
Для постановки диагноза основным является III этап диагностического поиска. Лабораторные исследования выявляют снижение гемоглобина в сочетании с низким цветовым показателем, ретикулоцитопе-нию. В сыворотке крови определяют высокое содержание железа, а в пунктате костного мозга — сидеробласты (клетки костного мозга с включениями железа в виде гранул). Дефицит ферментов, участвующих в обмене порфиринов, уточняют путем определения продуктов порфиринов в моче. Повышенное содержание железа в организме доказывается также с помощью десфераловой пробы (после введения десферала с мочой выделяется увеличенное количество железа). При биопсии печени, селезенки можно обнаружить признаки гемосидероза. ОЖСС у таких больных снижена.
Лечение.Назначение препаратов железа неэффективно, но еще больше увеличивает содержание железа в крови и способствует гемосидерозу органов. Точно так же не показаны и гемотрансфузии. Применяется пири-доксин (витамин В6) в дозах 50 — 200 мг/сут внутрь или по 100 мг внутримышечно 2 раза в неделю в течение 2 мес. Наиболее эффективен кофер-мент пиридоксаль-фосфат, так как иногда бывает блокирована возможность перехода пиридоксина в пиридоксаль-фосфат. При наследственных формах лечение витамином Вб надо повторять периодически. В случае ре-зистентности к терапии пиридоксином применяют анаболические и андро-генные гормоны.
Для уменьшения гемосидероза органов и снижения уровня сывороточного железа назначают десферал (внутривенно по 500 — 1000 мг) с перерывами, ориентируясь на уровень железа и присутствие сидероблас-тов в костном мозге.
Не нашли то, что искали? Воспользуйтесь поиском:
Лучшие изречения: При сдаче лабораторной работы, студент делает вид, что все знает; преподаватель делает вид, что верит ему. 8976 —  | 7163 —
| 7163 —  или читать все.
или читать все.
193.124.117.139 © studopedia.ru Не является автором материалов, которые размещены. Но предоставляет возможность бесплатного использования. Есть нарушение авторского права? Напишите нам | Обратная связь.
Отключите adBlock!
и обновите страницу (F5)
очень нужно
источник
Сидеробластная Анемия – это вид анемии, характеризующийся сниженным содержанием железа в эритроцитах из-за того, что костный мозг не использует его для синтеза гемоглобина. Вот что это такое и чем проявляется недуг, мы расскажем ниже.
Главными причинами возникновения болезни являются:
- наследственные факторы. Отмечается генетическая предрасположенность в 50% случаев. Болезнь сцеплена с Х-хромосомой или наследуется по аутосомно-доминантному типу;
- отравление свинцом, алкоголем, левомицетином;
- аутоиммунные заболевания;
- опухолевые процессы;
- лечение туберкулостатическими препаратами;
- пожилой возраст. У людей этой возрастной категории часто возникает идиопатическая приобретенная форма сидеробластной Анемии, причины которой до конца не установлены.
В основе патологического процесса, возникающего в организме из-за сидеробластной или сидероахристичекой Анемии, наблюдается нарушение процессов утилизации железа при нормальном его содержании в сыворотке крови. Это приводит к нарушению синтеза протопорфорина – вещества, которое соединяется с ионом железа и превращается в гем, входящий в состав гемоглобина.

В совокупности эти факторы приводят к образованию в костном мозге сидеробластов – клеток-предшественниц эритроцитов с вкраплениями гранул железа в виде кольца. В то же время происходит откладывание железа (гемосидероз) в таких органах, как печень, легкие, поджелудочная железа и сердце, что ведет к нарушению их функций.
Первые признаки наследственных форм появляются в детском возрасте и сопровождаются жалобами на слабость, головную боль, ухудшение памяти, повышенную утомляемость, невнимательность, ухудшение успеваемости в школе. При осмотре обнаруживают бледность кожных покровов, а также увеличение печени и селезенки.
Взрослые пациенты могут жаловаться на слабость, быструю утомляемость во время физических нагрузок. Обращает на себя внимание бледность кожи, которая может иметь землисто-серый оттенок, у трети больных возможен гепатолиенальный синдром. В анамнезе имеются факты контакта со свинцом, длительный прием лекарственных средств, сопутствующие болезни.
У всех категорий пациентов, по мере развития сидеробластной анемии, со временем наблюдаются признаки гемосидероза, который приводит к возникновению аритмии, сахарного диабета, легочной недостаточности.
Если вы заметили у себя симптомы болезни, следует срочно обратиться к врачу.
Для уточнения диагноза необходимо провести ряд анализов:
- общий анализ крови – покажет сниженный гемоглобин и цветной показатель, уменьшение количества ретикулоцитов;
- биохимический анализ крови укажет на нормальный или повышенный уровень сывороточного железа, высокие цифры ферритина;
- пункция костного мозга, в которой будут определяться сидеробласты;
- биопсия печени, в результате которой обнаружится накопление железа в клетках.
При возможности необходимо исключить факторы, способствующие хронической интоксикации (свинец, алкоголь).
В план лечения включают введение витамина В6 (пиридоксина) в течение двух месяцев. При устойчивой к пиридоксину форме назначают анаболические препараты, десферал для выведения излишков железа. Переливание крови используют крайне редко и в тяжелых случаях.
источник
Наследственные сидеробластные анемии — разнородная по спектру генетических нарушений и выраженности клинических проявлений группа заболеваний.
Наследственные сидеробластные анемии, сцепленные с Х-хромосомой. У большинства детей с сидеробластной анемией механизм наследования связан с Х-хромосомой. В связи с этим анемия диагностируется преимущественно у мальчиков и их близких родственников мужского пола по материнской линии (родные дяди и двоюродные братья). Редко имеется другой вариант наследования, не связанный с Х-хромосомой. В некоторых семьях заболевание возникает только у девочек, так как у мальчиков-гомозигот патология не совместима с жизнью.
В основе патогенеза лежит дефект синтеза 5-аминолевулинатсинтетазы (АЛК), участвующей в синтезе гема. Для нормальной работы этого фермента необходимо достаточное количество пиридоксина — витамина В6 (этим и объясняется эффективность лечения витамином В6). Активность фермента 5-аминолевулинатсинтетазы (АЛК-синтетазы) кодирует ген ALAS2, расположенный на Х-хромосоме. У больных сидеробластной анемией описано несколько различных мутаций данного гена.
Наследственные сидеробластные анемии с аутосомным типом наследования. Этот тип наследования встречается значительно реже, чем сцепленный с Х-хромосомой. Известны случаи как аутосомно-доминантного, так и аутосомно-рецессивного наследования. Мутации гена ALAS2 не определяются, поэтому терапевтический эффект пиридоксина отсутствует.
Спорадическая врожденная сидеробластная анемия. В мире описано около 20 случаев сидеробластной анемии, выявленной сразу после рождения, без признаков заболевания у других членов семьи. Вероятно, при этом имелись либо аутосомно-рецессивный тип наследования, либо появление новых мутаций гена ALAS2 в родительских половых клетках.
Митохондриальная цитопатия (синдром Пирсона). Синдром Пирсона — врожденное заболевание, которое обусловлено делециями или другими генетическими перестройками митохондриальной ДНК и клинически проявляется множественными органными поражениями. Одним из ранних признаков заболевания является тяжелая анемия, ассоциированная с наличием кольцевых сидеробластов в костном мозге. Продолжительность жизни детей с синдромом Пирсона обычно не превышает 2-3 года.
Синдром DIOMOAD. В основе большинства клинических проявлений заболевания, наследующегося по аутосомно-рецессивному типу, лежат дегенеративные процессы в нервной ткани, обусловленные, вероятно, наследственными дефектами метаболизма тиамина. Гематологическим проявлением синдрома является нормоцитарная сидеробластная анемия средней степени тяжести в сочетании с нейтропенией и выраженной тромбоцитопенией. Отмечен терапевтический эффект применения тиамина.
Классификация сидеробластных анемий 
В большинстве случаев клиническая картина сидеробластной анемии, сцепленной с Х-хромосомой, не отличается от таковой при анемии с аутосомным типом наследования или других врожденных форм. Тяжелая анемия, как правило, диагностируется в младенчестве или раннем детстве. При менее выраженных проявлениях анемического синдрома или бессимптомном течении заболевание выявляется обычно у взрослых или даже пожилых больных.
Наряду с анемическим синдромом у всех больных определяются признаки избытка железа в организме, которые получили специальное название: синдром эритропоэтигеского гемохроматоза. Проявления этого синдрома достаточно разнообразны. Чаще всего определяются умеренная гепатомегалия и спленомегалия. Функция печени в большинстве случаев нарушена незначительно или не страдает вообще. При биопсии печеночной ткани определяются депозиты железа в гепатоцитах. У некоторых больных старше 30-40 лет при гистологическом исследовании находят признаки микронодулярного цирроза печени, протекающего, как правило, доброкачественно.
На фоне гемохроматоза поджелудочной железы могут определяться сахарный диабет или нарушения толерантности к глюкозе. Редко при объективном обследовании выявляется пигментация кожных покровов. Наиболее опасными проявлениями синдрома эритро-поэтического гемохроматоза являются тяжелые нарушения сердечного ритма и сердечная недостаточность, которые развиваются, как правило, на поздних этапах заболевания. В тяжелых случаях у детей может наблюдаться задержка роста и развития.
В анализе крови определяется анемия различной степени тяжести. При тяжелой анемии обычно имеются гипохромия, микроцитоз, анизо- и пойкилоцитоз, реже мишеневидные клетки и единичные сидероциты. При менее тяжелых формах анемии в мазке могут выявляться две популяции клеток: гипохромные микроциты и нормальные эритроциты. На гистограмме эритроцитов при этом образуется двухфазная кривая, отражающая различия в размерах эритроцитов. Уровень лейкоцитов и тромбоцитов обычно в норме, но при развитии гиперспленизма может снижаться. Количество ретикулоцитов в большинстве случаев в норме или незначительно повышено.
В миелограмме выявляется гиперплазия эритроидного ростка на фоне нормобластического типа кроветворения и повышенное количество сидеробластов. В редких случаях (при сопутствующем дефиците фолиевой кислоты) может определяться мегалобластический тип кроветворения.
При биохимическом исследовании обычно выявляется незначительная гипербилирубинемия, повышение уровня ферритина и снижение уровня трансферрина, и увеличение его сатурации. При анемии, сцепленной с Х-хромосомой, имеется снижение активности АЛК-синтетазы.
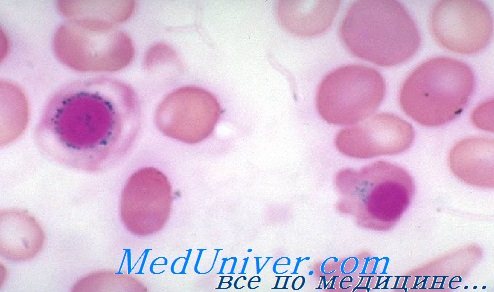 Мазок при сидеробластной анемии
Мазок при сидеробластной анемии
У больных с синдромом Пирсона диагностируется рефрактерная сидеробластная анемия, которая сочетается с признаками экзокринной недостаточности поджелудочной железы, эпизодами молочнокислого ацидоза и прогрессирующей почечной и печеночной недостаточностью. Анемия выявляется, как правило, после рождения и носит нормоцитарныи или макроцитарный характер. Уровень ретикулоцитов чаще всего снижен. В большинстве случаев имеются нейтропения и тромбоцитопения различной степени тяжести. При электрофорезе гемоглобина обычно определяется повышение уровня Hb F. Костный мозг гиперклеточный или нормоклеточный, выявляются кольцевые сидеробласты.
Всем больным наследственными сидеробластными анемиями необходимо начинать терапию пиридоксином (витамин В6), которая эффективна в среднем в 1/3 случаев. Доза витамина В6 обычно составляет 50-100 мг в сутки. Выраженность терапевтического эффекта у пациентов, ответивших на лечение, различна. В большинстве случаев появляется ретикулоцитоз и уровень гемоглобина в течение 1-2 месяцев лечения постепенно повышается до нормы или субнормальных значений. Морфологические изменения эритроцитов при этом сохраняются.
При отсутствии оптимального эффекта на фоне терапии пиридоксином уровень гемоглобина стабилизируется, но не достигает нормальных значений. Необходимо проведение поддерживающей терапии витамином В6, при отсутствие которой концентрация гемоглобина через несколько месяцев снижается до исходных величин. При выявлении мегалобластического типа кроветворения показано лечение фолиевой кислотой. Пациентам с тяжелой анемией, у которых отсутствует эффект витамина В6, по показаниям проводятся периодические трансфузии эритроцитарной массы. Это уменьшает выраженность анемии и предотвращает задержку роста и развития у детей.
Для профилактики гемосидероза показана терапия дефероксамином, в которой нуждаются прежде всего больные с тяжелой анемией, получающие гемотрансфузионную терапию. Предпочтительно проведение 12-часовых подкожных инфузий препарата в дозе 40 мг/(кг-день) каждые 5 дней недели (этот режим обладает минимальной токсичностью). Спленэктомия при наследственных формах сидеробластной анемии довольно часто осложняется тромбоэмболией, в ряде случаев с летальным исходом, поэтому оперативное лечение используется редко.
источник
Сидероахрестическая анемия (САА) — железонасыщенная или сидеробластная анемия, при которой эритроциты содержат мало железа (гипохромны) вследствие его неиспользования костным мозгом для синтеза гемоглобина.
В основе развития сидероахрестических анемий лежит нарушение синтеза гема. Железо и белок, необходимые для синтеза гемоглобина, есть, но отсутствует достаточное количество протопорфирина. Вследствие этого не происходит синтез гема — основного компонента молекулы гемоглобина. Гем — соединение порфириновых колец (протопорфирина) с атомом железа. Соединяясь с глобином, он образует молекулу гемоглобина.
При САА уменьшается образование порфиринов и возникает избыток железа. Уменьшение образования первых обусловлено врожденным или приобретенным дефицитом ряда ферментов. Накопление железа в организме приводит к его отложению во внутренних органах.
Выделяют две основные наследственные формы САА: пиридоксинзависимую (дефицит пиридоксальфосфата, в связи с чем эффективно назначение пиридоксина) и пиридоксинрезистентную (регистрируют крайне редко). Существует непосредственный ферментный дефект (дефицит гемсинтетазы, обеспечивающей включение железа в молекулу гема).
Приобретенные формы чаще обнаруживают в пожилом возрасте, заболевание не носит семейного характера. САА чаще возникает при лечении туберкулостатическими препаратами, приводящем к истощению запасов пиридоксальфосфата, свинцовой интоксикации и алкоголизме. Возможно развитие идиопатических форм САА.
При наследственных формах заболевание начинается уже в раннем детстве . На первом этапе диагностического поиска обнаруживают жалобы, обусловленные циркуляторно-гипоксическим синдромом. В анамнезе — указания на бледность, слабость, увеличение печени и селезенки. Дети быстро устают, плохо учатся и имеют плохую память. Взрослые отмечают слабость
и снижение толерантности к физической нагрузке, возникающее после длительного лечения основного заболевания (туберкулеза) и воздействия профессиональных вредностей (контакт со свинцом). В анамнезе есть указания на обнаружение низкой концентрации гемоглобина и неэффективное лечение препаратами железа.
На втором этапе диагностического поиска в период обострения можно обнаружить бледность кожного покрова и видимых слизистых оболочек, у части больных — умеренное увеличение печени и селезенки. В связи с этим у таких пациентов обычно предполагают хроническое заболевание печени (чаще всего — хронический гепатит).
Отложение железа во внутренних органах может привести к возникновению ряда своеобразных симптомов. Так, отложение железа в поджелудочной железе вызывает развитие сахарного диабета, в печени — цирроза печени, в сердце — сердечной недостаточности, в половых железах — возникновение евнухоидизма.
Основным в установлении диагноза считают третий этап диагностического поиска. При лабораторном исследовании обнаруживают снижение концентрации гемоглобина в сочетании с низким цветовым показателем и нормальным или повышенным количеством ретикулоцитов. В крови определяют высокое содержание железа, а в пунктате костного мозга — повышенное количество сидеробластов (клетки костного мозга с включениями железа в виде гранул). Дефицит ферментов, участвующих в обмене порфиринов, уточняют посредством определения содержания продуктов распада последних в моче. Повышение концентрации железа в организме подтверждают с помощью пробы с дефероксамином (после введения препарата с мочой выделяется повышенное количество железа). При биопсии печени и селезенки можно обнаружить признаки гемосидероза. Общая железосвязывающая способность сыворотки у таких больных снижена.
Назначение препаратов железа неэффективно и еще больше увеличивает содержание железа в крови, способствуя гемосидерозу органов. Проведение гемотрансфузий не рекомендовано. Назначают прием внутрь пиридоксина в дозе 50-200 мг/сут или его внутримышечное введение по 100 мг 2 раза в неделю на протяжении 2 мес. Наиболее эффективно использование кофермента пиридоксаль фосфата, так как иногда блокируется возможность трансформации пиридоксина в пиридоксальфосфат. При наследственных формах лечение пиридоксином необходимо периодически повторять.
Для уменьшения гемосидероза органов и снижения концентрации сывороточного железа, ориентируясь на его содержание и присутствие сидеробластов в костном мозге, назначают дефероксамин (внутривенно по 500-1000 мг с перерывами).
Сущность В12-дефицитной анемии (В12ДА) состоит в нарушении образования ДНК и РНК в связи с нехваткой в организме витамина В12 (цианокобаламина), что приводит к нарушению кроветворения, появлению в костном мозге мегалобластов, внутрикостномозговому разрушению эритрокариоцитов, снижению количества эритроцитов и гемоглобина, лейкопении, нейтропении и тромбоцитопении, а также к изменениям в ряде органов и систем (ЖКТ, ЦНС).
В12ДА регистрируют значительно реже, чем ЖДА. Дефицит витамина В12 в организме может носить либо приобретенный, либо наследственный характер, т.е. быть генетически обусловленным.
Ниже перечислены причины развития В12ДА.
• Нарушения всасывания витамина В12.
• Приобретенные формы дефицита витамина В12:
— нарушение секреции гастромукопротеина (внутреннего фактора) в желудке;
— атрофия париетальных клеток слизистой оболочки желудка;
— антитела к париетальным клеткам слизистой оболочки желудка;
— антитела к гастромукопротеину или комплексу «гастромукопротеин + витамин В12»;
— органические поражения желудка (гастрэктомия, опухоли желудка, распространенный полипоз желудка);
— органические заболевания тонкой кишки (резекция кишечника, илеит, БК, спру).
• Наследственные формы дефицита витамина В12:
— наследственный дефицит внутреннего фактора (гастромукопротеина);
— генетически обусловленные нарушения всасывания комплекса «гастромукопротеин + витамин В12» в энтероцитах (болезнь ИмерслундГресбека);
— наследственный дефицит и функциональные аномалии транскобаламина II.
• Повышенный расход витамина В12:
— изменения кишечной микрофлоры при дивертикулезе кишечника;
• Уменьшенное потребление витамина В12:
— отсутствие в рационе продуктов животного происхождения;
Сходную с В12ДА гиперхромную анемию вызывает дефицит фолиевой кислоты, который возникает:
• при ее повышенном расходе (беременность);
• вскармливании детей козьим молоком;
• нарушении всасывания (органические заболевания кишечника, алкоголизм);
• приеме некоторых лекарственных препаратов (противосудорожные, противотуберкулезные средства, фенобарбитал, контрацептивы и др.).
Витамин B12 (цианокобаламин) не синтезируется в организме человека и поступает только с продуктами питания. Больше всего его содержится в мясе, яйцах, молоке, сыре, печени и почках. Запасы витамина B12 у взрослого человека достаточно велики, составляют 2-5 мг и преимущественно находятся в печени. Для развития дефицита цианокобаламина вследствие нарушений всасывания либо поступления витамина B12 с пищей требуется от 6 мес до 3-5 лет.
Чистый витамин B12 не способен всасываться в кишечнике. Для всасывания в подвздошной кишке цианокобаламин должен предварительно соединиться с так называемым внутренним фактором — гликопротеином (гастромукопротеин), синтезируемым париетальными клетками слизистой оболочки желудка.
Перенос всосавшегося витамина B12 кровью в костный мозг для участия в кроветворении осуществляется с помощью специфических транспортных белков — транскобаламинов I, II и III.
Витамин B12 состоит из двух коферментов — метилкобаламина и дезоксиаденозилкобаламина. Дефицит первого кофермента обусловливает нарушение синтеза ДНК, вследствие чего нарушается деление и созревание клеток красного ряда и происходит их избыточный рост без утраты ядра. Большие клетки, содержащие ядра, называют мегалобластами. Они не созревают до мегалоцитов (гигантские эритроциты без ядер) и легко гемолизируются, еще находясь в костном мозге. Дефицит витамина B12 вызывает нарушение роста клеток лейкоцитарного и тромбоцитарного ряда, но это не так заметно сказывается на их морфологии и количестве клеток, как нарушения эритропоэза.
При недостатке второго кофермента нарушается обмен жирных кислот, вследствие чего в организме происходит накопление токсичных продуктов — пропионовой и метилмалоновой кислоты: развивается поражение заднебоковых канатиков спинного мозга — фуникулярный миелоз (рис. 5-3).
Клиническая картина В12ДА, как это вытекает из схемы патогенеза, складывается из следующих синдромов:
• циркуляторно-гипоксического (при достаточной выраженности анемии и кислородного голодания тканей);
• гематологического (анемия гиперхромного типа).
Кроме этих синдромов, клиническую картину также определяет заболевание, на основе которого развилась В12ДА.
На первом этапе диагностического поиска при достаточно выраженной анемии могут возникать симптомы, обусловленные циркуляторно-
Рис. 5-3. Патогенез В12-дефицитной анемии
гипоксическим синдромом (слабость, повышенная утомляемость, одышка при физической нагрузке, боли в области сердца, сердцебиение). В случае нерезкого кислородного голодания тканей эти жалобы могут отсутствовать. Снижение аппетита, отвращение к мясу, боли и жжение в кончике языка, чувство тяжести в эпигастральной области после приема пищи, а также чередование поноса и запора обусловлены поражением ЖКТ и, в частности, выраженной секреторной недостаточностью желудка. При поражении ЦНС больные жалуются на головную боль, неустойчивую походку, зябкость, чувство онемения в конечностях и ощущение «ползания мурашек». Выраженность этих жалоб не всегда соответствует степени анемии. В период ремиссии заболевания они могут отсутствовать. Весьма существенно, если все перечисленные жалобы предъявляет немолодой человек; в таких случаях вероятность обнаружения В12ДА повышается.
В семейном анамнезе у больных с предполагаемой В12ДА могут быть указания на это заболевание среди родственников. Одна из причин развития анемии — злоупотребление алкоголем.
Данные анамнеза могут помочь в определении патогенетического варианта анемии. Ее развитие после пребывания больного возле больших водоемов и употребления в пищу сырой или недостаточно обработанной рыбы заставляет предположить в качестве возможной причины дифиллоботриоз. Если заболевание возникло у пожилого человека, страдающего хроническим гастритом, и развивается медленно, то можно думать о В12ДА. Если нарушения со стороны ЖКТ сочетаются со снижением массы тела и быстро прогрессируют, то в ка-
честве причины заболевания следует предположить злокачественное новообразование.
Наконец, сведения об успешном лечении больного цианокобаламином позволяют с большой уверенностью рассматривать имеющиеся признаки как симптомы В12ДА.
На втором этапе диагностического поиска симптомы могут быть обусловлены поражением ЖКТ и ЦНС. Кроме того, вероятность существования у пациента В12ДА повышает обнаружение ряда неспецифических признаков. Так, при В12ДА отмечают бледность кожного покрова в сочетании с небольшой иктеричностью склер и одутловатостью лица. Масса тела таких больных, как правило, нормальная или повышенная. При ее снижении в качестве возможной причины В12ДА нередко рассматривают злокачественную опухоль. Аналогичное значение имеет обнаружение увеличенного плотного лимфатического узла (возможно, метастаза опухоли). Циркуляторно-гипоксический синдром манифестирует так же, как и при ЖДА (расширение границ сердца влево, тахикардия, систолический шум, шум «волчка» на яремных венах).
Несомненное диагностическое значение имеет обнаружение при обследовании признаков глоссита (сглаженные сосочки языка вплоть до их полной атрофии — «полированный» язык). Печень несколько увеличена, можно пропальпировать селезенку. Тем не менее все эти симптомы не обязательны для В12ДА. Отмечают нарушения глубокой чувствительности, нижний спастический парапарез (картина псевдотабеса). Следует отметить, что нарушения со стороны нервной системы возникают далеко не во всех случаях, поэтому их отсутствие не исключает существования В12ДА.
Таким образом, результаты второго этапа в сочетании с анамнестическими данными и жалобами больного дают основание лишь заподозрить В12ДА. Окончательный диагноз ставят после проведения серии лабораторных исследований.
На третьем этапе диагностического поиска при исследовании периферической крови обнаруживают следующие изменения: снижение количества эритроцитов (менее 3х1012/л), повышение цветового показателя (более 1,1), среднего содержания гемоглобина в эритроците (более 34 пг) и среднего объема эритроцита (более 120 мкм3). Эритроцитометрическая кривая сдвинута вправо — увеличено количество макроцитов, присутствуют мегалоциты (эритроциты диаметром более 12 мкм). Форма эритроцитов изменена (пойкилоцитоз). Отмечают единичные мегалобласты.
Дополнительный признак — присутствие нейтрофилов с гиперсегментированными ядрами.
Если в картине периферической крови не обнаруживают характерных признаков, то выполняют стернальную пункцию. Последняя позволяет обнаружить в костном мозге мегалобластический тип кроветворения.
Важным считают определение содержания сывороточного железа: при В12ДА оно может соответствовать норме или быть повышенным в связи с усиленным гемолизом эритроцитов. В этих случаях увеличено содержание непрямого билирубина. При исследовании желудочного сока часто обнаруживают гистами-
ноустойчивую ахилию (характерный признак анемии Аддисона-Бирмера), а эндоскопически — атрофию слизистой оболочки желудка.
Другие инструментальные методы исследования помогают обнаружить признаки миокардиодистрофии (развивается на фоне выраженной анемии), а также уточнить этиологию заболевания.
В диагностике В12ДА выделяют два этапа:
1) доказательство дефицита витамина В12 в качестве причины анемии;
2) установление причин дефицита витамина В12. Критерии В12ДА:
• снижение содержания эритроцитов (менее 3,0х1012/л);
• повышение цветового показателя (более 1,1);
• повышение содержания гемоглобина в эритроцитах (более 34 пг);
• увеличение среднего объема эритроцита (более 120 мкм3);
• сдвиг эритроцитометрической кривой вправо (увеличение количества макроцитов, присутствие мегалоцитов — эритроцитов диаметром более 12 мкм);
• обнаружение в мазках пунктата костного мозга элементов мегалобластного кроветворения;
• повышение содержания сывороточного железа более 30,4 мкмоль/л;
• снижение радиоактивности мочи после приема витамина B12, меченного радиоактивным кобальтом.
Для установления причины анемии следует проводить рентгенологическое, эндоскопическое (опухоль желудка, дивертикулез тонкой кишки), гельминтологическое исследование (инвазия широким лентецом), функциональное исследование печени с биопсией (хронический гепатит, цирроз) и определение нейтрального жира в кишечнике (спру).
В12ДА следует дифференцировать от фолиеводефицитной анемии. При дефиците фолиевой кислоты обнаруживают макроцитарную гиперхромную анемию, а в костном мозге — мегалобласты. Следует отметить, что дефицит фолиевой кислоты регистрируют значительно реже. В отличие от В12ДА, при фолиеводефицитной анемии содержание фолиевой кислоты в крови и эритроцитах снижено. Кроме того, при воздействии на препарат костного мозга ализарином красным окрашиваются только мегалобласты, связанные с дефицитом витамина В12, и не окрашиваются мегалобласты, связанные с дефицитом фолиевой кислоты.
Возможно резкое обострение заболевания. В таких случаях развивается коматозное состояние: потеря сознания, снижение температуры тела и АД, одышка, рвота, арефлексия и непроизвольное мочеиспускание. Между развитием коматозного состояния и снижением концентрации гемоглобина нет четкой зависимости (у больных с его резко сниженным содержанием кома отсутствует). Главную роль в патогенезе комы играют быстрый темп и степень снижения гемоглобина, а также резкая ишемия и гипоксия ЦНС.
Формулировка развернутого клинического диагноза должна учитывать:
• этиологию В12ДА (отдельно следует выделять анемию, вызванную болезнью Аддисона-Бирмера);
• стадию процесса (рецидив или ремиссия);
• выраженность отдельных синдромов (как правило, при неврологических расстройствах, обусловленных фуникулярным миелозом).
Комплекс лечебных мероприятий при В12ДА следует проводить с учетом этиологии, выраженности анемии и существования неврологических нарушений. При лечении следует ориентироваться на следующие положения.
• Непременное условие лечения В12ДА при глистной инвазии — дегельминтизация (для изгнания широкого лентеца по определенной схеме назначают никлозамид или папоротника мужского корневища).
• При органических заболеваниях кишечника и поносе следует применять ферментные препараты (панкреатин, гемицеллюлаза + желчи компоненты + панкреатин), а также закрепляющие средства (карбонат кальция в сочетании с дерматолом).
• Нормализации кишечной микрофлоры достигают приемом ферментных препаратов (панкреатин, гемицеллюлаза + желчи компоненты + панкреатин), а также подбором диеты, способствующей ликвидации синдромов гнилостной или бродильной диспепсии.
• Сбалансированное питание с достаточным содержанием витаминов белка и безусловным запрещением употребления алкоголя — непременное условие лечения В12ДА и фолиеводефицитной анемии.
• Патогенетическое лечение осуществляют с помощью парентерального введения цианокобаламина (ликвидация его дефицита), нормализации показателей центральной гемодинамики и нейтрализации антител к гастромукопротеину (внутреннему фактору) или комплексу гастромукопротеин + витамин В12 (применение глюкокортикоидов). Цианокобаламин вводят внутримышечно в дозе 200-500 мкг 1 раз в день ежедневно в течение 4-6 нед до наступления гематологической ремиссии. Критерии последней: резкое увеличение количества ретикулоцитов в периферической крови (ретикулоцитарный криз) и трансформация мегалобластического кроветворения в нормобластическое. Развитие ретикулоцитарного криза на 5-6-й день лечения — ранний критерий его эффективности. В процессе лечения цианокобаламином количество эритроцитов увеличивается быстрее, чем содержание гемоглобина, поэтому цветовой показатель обычно снижается. После нормализации костномозгового кроветворения и состава крови (обычно — через 1,5-2 мес) препарат вводят 1 раз в неделю в течение 2-3 мес, а затем — 2 раза в месяц в тех же дозах, что и в начале курса, в течение полугода). В дальнейшем больных ставят на диспансерный учет и с профилактической целью вводят им цианокобаламин (1-2 раза в год короткими курсами по 5-6 инъекций или ежемесячно по 200-500 мкг пожизненно).
При симптомах фуникулярного миелоза в значительных дозах вводят цианокобаламин (по 500-1000 мкг ежедневно в течение 10 дней, а затем 1-3 раза в неделю до исчезновения неврологических нарушений).
Гемотрансфузии проводят лишь при значительном снижении гемоглобина и возникновении симптомов коматозного состояния. Рекомендовано введение эритроцитарной массы в дозе 250-300 мл (5-6 трансфузий).
Преднизолон (по 20-30 мг/сут) назначают при заболевании аутоиммунной этиологии.
В настоящее время применение цианокобаламина сделало прогноз В12ДА благоприятным. При полноценном лечении больные живут длительное время.
Мер первичной профилактики не существует. У лиц, имеющих ранее перечисленные этиологические факторы, следует периодически исследовать кровь для своевременного обнаружения анемии.
Гемолитические анемии (ГА) — обширная группа заболеваний, значительно различающихся по этиологии, патогенезу, клинической картине и лечению. Основной патологический процесс, объединяющий эти заболевания в одну группу, — повышенный гемолиз. Он может происходить внутриклеточно (в макрофагах селезенки, как обычный физиологический процесс) и непосредственно в сосудах (внутрисосудистый или внеклеточный гемолиз). В норме продолжительность жизни эритроцита составляет 100-120 дней, но при гемолитических анемиях она сокращается до 12-14 дней.
Усиленный гемолиз, происходящий в клетках мононуклеарно-фагоцитарной системы (главным образом в селезенке), манифестирует следующими симптомами:
• в крови увеличивается содержание свободного (непрямого) билирубина, с чем связано желтушное окрашивание кожи и слизистых оболочек разной степени выраженности;
• гепатоциты перерабатывают избыточное количество непрямого билирубина в прямой билирубин, вследствие чего возникает интенсивное окрашивание желчи (плейохромия) и развивается склонность к образованию камней в желчном пузыре и протоках;
• в кишечнике, куда поступает желчь, в большом количестве образуется стеркобилиноген и уробилиноген, в связи с чем возникает интенсивное окрашивание каловых масс;
• в моче увеличивается содержание уробилина;
• общее количество эритроцитов уменьшается, увеличивается число ретикулоцитов в периферической крови и содержание эритробластов и нормоцитов в костном мозге.
Признаки усиленного внутрисосудистого гемолиза:
• увеличение концентрации свободного гемоглобина в крови;
• выделение свободного гемоглобина с мочой в неизмененном виде или в виде гемосидерина (моча красного, бурого или почти черного цвета);
• отложение гемосидерина во внутренних органах (гемосидероз).
Все гемолитические анемии разделяют на две большие группы — наследственные и приобретенные. Наследственные ГА обусловлены генетическими дефектами эритроцитов, которые становятся функционально неполноценными и легко разрушаются. Приобретенные ГА — следствие воздействия на нормальные эритроциты различных факторов (образование антител, гемолитических ядов, механические воздействия и др.), приводящих к их разрушению.
— связанные с нарушением мембраны эритроцитов (гемолитическая микросфероцитарная анемия или болезнь Минковского-Шоффара, овалоцитоз, стоматоцитоз);
— связанные с нарушением активности ферментов (глюкозо-6- фосфатдегидрогеназы (Г-6-ФД), пируваткиназы, глутатионредуктазы и др.) в эритроцитах;
— связанные с нарушением структуры или синтеза цепей глобина (талассемия, серповидноклеточная анемия и др.).
— связанные с воздействием антител (изоиммунные, аутоиммунные);
— связанные с изменением структуры мембраны эритроцитов вследствие соматической мутации (пароксизмальная ночная гемоглобинурия или болезнь Маркиафавы-Микели);
— связанные с механическим повреждением мембраны эритроцита (протезы клапанов сердца, маршевая гемоглобинурия);
— обусловленные химическими повреждениями эритроцитов (гемолитические яды, свинец, тяжелые металлы, органические кислоты);
— обусловленные недостатком витамина Е;
— связанные с воздействием паразитов (малярия).
Частота возникновения тех или иных ГА весьма различна. Так, наследственный микросфероцитоз в Европе регистрируют с частотой 0,03\%, а в Японии и Африке значительно реже. Частота развития анемии, обусловленной дефицитом Г-6-ФД, высока в странах средиземноморского бассейна, на Ближнем Востоке и на Кавказе. Ночная пароксизмальная гемоглобинурия — редкое заболевание. В дальнейшем будет рассмотрена диагностика трех видов наиболее распространенных ГА: наследственного микросфероцитоза, талассемии и аутоиммунной ГА.
Наследственный микросфероцитоз (болезнь Минковского-Шоффара)
В основе микросфероцитарной ГА (наследственного микросфероцитоза) лежит дефект оболочки эритроцита, наследуемый по аутосомно-доминантному типу. Сферические ригидные эритроциты не могут менять форму при продвижении по узким капиллярам, особенно в синусах селезенки, в связи с чем теряется часть оболочки эритроцита, и происходит гемолиз. Мембрана эритроцита пропускает внутрь повышенное количество ионов натрия, накопление которых способствует усилению расхода АТФ и глюкозы на последующее выведение их из клетки. Это также приводит к укорочению продолжительности жизни эритроцита.
Клиническая картина заболевания определяется гемолитическим синдромом и сопутствующими врожденными аномалиями скелета и внутренних органов . Болезнь течет волнообразно: «спокойные» периоды прерывают гемолитические кризы, провоцируемые развитием неспецифических инфекционных поражений, во время которых гемолиз резко интенсифицируется и все симптомы болезни усиливаются.
На первом этапе диагностического поиска можно получить информацию о жалобах больного на периодически возникающую легкую желтушность кожного покрова и преходящую слабость. При тяжелом течении заболевания регистрируют гемолитические кризы, обычно возникающие спонтанно либо под влиянием инфекционных возбудителей, переутомления, травмы и переохлаждения; отмечают озноб, повышение температуры тела, боли в мышцах, в области печени и селезенки. Резко усиливается желтуха, моча и кал темнеют. Если симптомы криза выражены не столь резко, но желтуха выражена вполне отчетливо, то таких больных нередко госпитализируют в инфекционные больницы с подозрением на вирусный гепатит, где обычно диагноз не подтверждают. Постоянная желтушность вне кризов у таких больных может послужить основанием для предположения о хроническом гепатите.
На втором этапе диагностического поиска обнаруживают лимонножелтое окрашивание кожи, усиливающееся до более интенсивной желтухи в период гемолитического криза. У части больных можно отметить врожденные аномалии (башенный череп, заячья губа, пороки сердца). При выраженной анемии регистрируют циркуляторно-гипоксический синдром (анемический систолический шум, тахикардия, снижение АД, шум «волчка» на яремных венах и др.). Гемолиз происходит в селезенке, поэтому со временем орган увеличивается. Данные второго этапа диагностического поиска скорее исключают ряд заболеваний печени, способных стать причиной желтухи, а не подтверждают наследственный микросфероцитоз.
Решающим считают третий этап диагностического поиска, во время которого обнаруживают синдром гемолиза, протекающий у больных наследственным микросфероцитозом с некоторыми особенностями.
Общий анализ крови позволяет определить снижение содержания гемоглобина и эритроцитов. Основной морфологический признак заболевания — присутствие в крови большого количества мелких круглых эритроцитов (микросфероцитов). Их диаметр уменьшен, а осмотическая резистентность значительно снижена. Гемолиз начинается при концентрации хлорида натрия 0,6-0,8\%, а при его содержании около 0,4\% происходит полный гемолиз. В норме же он начинается при концентрации 0,42-0,46\%, а становится полным при содержании хлорида натрия около 0,30-0,32\%.
Усилен аутогемолиз: во время инкубации эритроцитов в течение 48 ч при температуре 37 °С гемолизируется не менее 30\%, тогда как в норме — лишь 3-4\% клеток. Положительны пробы с АТФ и дектрозой: их добавление к эритроцитам уменьшает аутогемолиз. Продолжительность жизни эритроцита, определяемая с помощью эритроцитов, меченных 51Сr, при наследственном микросфероцитозе укорочена.
В крови определяют и другие признаки гемолиза: ретикулоцитоз и увеличение концентрации непрямого билирубина. В кале повышено содержание стеркобилина, а в моче — уробилина. При длительном течении болезни холецистография и УЗИ позволяют обнаружить в желчном пузыре и протоках конкременты.
• камнеобразование в желчном пузыре;
• гипергенераторная анемия и желтуха в период криза;
• микросфероциты в мазке крови;
• снижение осмотической резистентности эритроцитов после инкубации цельной крови в стерильных условиях в течение суток при 37 °С в случаях, когда количество сфероцитов превышает 1-2\% общего числа эритроцитов;
• усиление спонтанного гемолиза после 49-часовой инкубации крови в стерильных условиях до 10-50\% (в норме лизируется менее 4\%), при этом аутогемолиз удается предотвратить добавлением к эритроцитам до начала инкубирования декстрозы.
Таким образом, как и при других видах анемии, диагностика заболевания основана преимущественно на данных третьего этапа, но и результаты второго этапа поиска имеют значение.
Формулировку развернутого клинического диагноза осуществляют в следующей последовательности:
• фаза (обострение (гемолитический криз) или ремиссия);
• состояние внутренних органов (спленомегалия, желчнокаменная болезнь, возможные аномалии скелета и других органов).
Единственный эффективный метод лечения — спленэктомия, после чего патологический гемолиз прекращается, хотя эритроциты и имеют дефектную
оболочку. Выполнение этого вмешательства рекомендовано при тяжелом течении болезни и частых гемолитических кризах. При резкой анемии допустимо переливание эритроцитарной массы.
Препараты железа, цианокобаламин и глюкокортикоиды применять не следует вследствие их неэффективности (механизмы развития анемии не связаны с дефицитом железа и витамина В12, а гемолиз не связан с противоэритроцитарными антителами).
В основе талассемии лежит нарушение синтеза одного вида цепей глобина, что связано с наследственным дефектом транспортной РНК или гена-регулятора. Может быть нарушен синтез различных цепей глобина, в связи с чем различают α-, β- и γ-талассемию. Возможно сочетание нарушения синтеза различных цепей. Чаще всего обнаруживают изменения β-цепей, в связи с этим содержание нормального гемоглобина А, в состав которого входят две α-цепи и две β-цепи, уменьшается, а концентрация гемоглобина F и А2 увеличивается. Эритроциты, содержащие аномальный гемоглобин, легко разрушаются, секвестрируются и гемолизируются в узких капиллярах селезенки. Этому способствует повышение проницаемости мембраны эритроцита.
Различают два варианта течения β-талассемии:
• большая талассемия (анемия Кули), регистрируемая в детском возрасте (гомозиготная форма);
• малая талассемия, регистрируемая у взрослых (гетерозиготная форма). Ниже рассмотрены этапы диагностического поиска при малой талассемии. На первом этапе диагностического поиска можно обнаружить жалобы
на повышенную утомляемость, слабость и головные боли. Они неспецифичны для этого вида ГА, как и преходящая желтушность, которая тем не менее должна обратить на себя внимание врача. В анамнезе — указания на периоды повышения концентрации билирубина, обнаружение увеличенной селезенки, а также безуспешность лечения препаратами железа. Такие же симптомы могут быть и у родственников больного, что указывает на наследственный характер заболевания.
На втором этапе диагностического поиска в периоды обострения возможно обнаружение умеренной желтушности кожного покрова, а у половины больных — увеличение селезенки. В связи с этим у таких пациентов предполагают существование хронического заболевания печени (чаще всего — хронического гепатита).
Основным для установления диагноза считают третий этап диагностического поиска. При лабораторном исследовании обнаруживают признаки гемолиза (увеличение содержания непрямого билирубина и ретикулоцитов, уробилинурию), а также характерные симптомы талассемии:
• гипохромную анемию в сочетании с высокой концентрацией сывороточного железа;
• характерные мишеневидные эритроциты;
• увеличение числа малых фракций гемоглобина (гемоглобин А2 и F).
Диагностические критерии талассемии:
• выраженная гемолитическая анемия с гипохромными микроцитарными эритроцитами;
• анизоцитоз, множество мишеневидных, а также капле- и сигарообразных эритроцитов в мазке периферической крови;
• увеличение фракции гемоглобина F, отсутствие фракции гемоглобина А;
• возможно увеличение фракции гемоглобина А2 почти в 2 раза.
• Гемотрансфузии + прием дефероксамина (во избежание развития гемосидероза) .
• Прием фолиевой кислоты по 0,005 г 1-2 раза в день при снижении концентрации гемоглобина в связи с инфекционными заболеваниями или во время беременности.
• Прием витаминов группы В (В6, B12) и аскорбиновой кислоты для улучшения эритропоэза (подобное назначение небесспорно).
• Спленэктомия при значительной спленомегалии и распаде эритроцитов в селезенке.
Аутоиммунная гемолитическая анемия
Аутоиммунная гемолитическая анемия (АИГА) — распространенная форма приобретенных ГА. Выделяют два варианта болезни:
• симптоматическую форму, при которой анемия развивается на фоне определенного заболевания (гемобластоза, системного заболевания соединительной ткани, хронического активного гепатита, опухоли, НЯК и др.);
• идиопатическую форму, когда обнаружить определенное заболевание не удается (острое инфекционное заболевание, беременность, роды и травма в анамнезе не служат причиной АИГА, а лишь провоцируют ее обострение).
При АИГА вырабатываются антитела к собственному антигену эритроцитов. Первый этап патогенеза АИГА — изменение антигена эритроцитов под влиянием лекарственных препаратов, вирусов или бактерий. Возможна также соматическая мутация единичного иммуноцита. Дальнейшая реакция антител и антигенов эритроцитов обусловливает развитие гемолиза и анемии.
АИГА может развиваться при участии различного вида аутоантител, вызывающих гемолиз при различной температуре. Различают два типа антител — тепловые (реагируют с эритроцитами при температуре тела не ниже 37 °С) и холодовые (реагируют с эритроцитами при температуре ниже 37 °С). На этом основании выделяют четыре вида АИГА:
• АИГА с неполными тепловыми агглютининами;
• АИГА с тепловыми гемолизинами;
• АИГА с холодовыми агглютининами;
• АИГА с двухфазными гемолизинами.
Образующийся комплекс «эритроцит + антитело» поглощают макрофаги селезенки (внутриклеточный гемолиз). Также могут вырабатываться аутоиммунные антитела к тромбоцитам, что приводит к развитию тромбоцитопении.
Чаще всего регистрируют АИГА, обусловленные тепловыми аутоантителами. Последние принадлежат к IgG и служат неполными тепловыми агглютининами, максимально демонстрирующими свое действие при температуре 37 °С. Гемолиз происходит внутриклеточно и существенно реже — внутри сосудов.
Холодовые аутоантитела принадлежат к IgM и представлены агглютининами. Гемолиз возникает в результате их соединения с эритроцитами и комплементом. Действие антител начинается при низкой температуре (ниже 32 °С): в мелких сосудах дистальных отделов тела (пальцы рук, ног, кончики ушей и носа) образуются крупные конгломераты из агглютинированных эритроцитов, а сами сосуды спазмируются. Гемолиз происходит преимущественно внутриклеточно, но обнаружение гемоглобинурии указывает и на внутрисосудистый гемолиз. При переходе пациента в теплое помещение гемолиз прекращается.
Значительно реже возникают АИГА, обусловленные действием двух других типов аутоантител — тепловых и двухфазных холодовых гемолизинов. При обоих вариантах агглютинации эритроцитов не происходит. Гемолиз возникает при осаждении аутоантител (гемолизинов) на эритроцитах, в связи с чем он происходит внутри сосудов и сопровождается выделением черной мочи (гемоглобинурия).
Под действием тепловых гемолизинов гемолиз происходит в обычных условиях (пребывание на холоде — не обязательное условие). Во время пребывания больного на холоде двухфазные гемолизины осаждаются на эритроцитах, но собственно гемолиз начинается лишь после перехода больного в теплое помещение.
Клиническая картина АИГА полиморфна и обусловлена:
• быстротой развития гемолиза (кризовое или более спокойное течение);
• преобладающим патогенетическим механизмом гемолиза (те или иные аутоантитела приводят к гемолизу при различных внешних условиях);
• изменениями в органах (в частности, в печени и селезенке);
• местом, где происходит гемолиз (селезенка, сосудистое русло);
• фоновыми заболеваниями (при вторичных АИГА).
В связи с этим при конечном сходном результате — гемолизе эритроцитов и развитии всех признаков ГА — на всех трех этапах диагностического поиска можно получить совершенно различные данные.
Ha первом этапе диагностического поиска больные с гемолитическими кризами, обычно развивающимися после травм и инфекционных заболеваний, предъявляют жалобы на повышение температуры тела, боли в пояснице, озноб и возникновение желтушности. При АИГА, спровоцированных воздействием холода, отмечают непереносимость низких температур: у больных синеют дис-
тальные участки конечностей, нос и уши. Как правило, они плохо себя чувствуют в холодное время года.
На втором этапе диагностического поиска (без учета симптомов основного заболевания при вторичных формах АИГА) обычно возникают две ситуации:
• в период ремиссии, кроме легкой желтушности и незначительного увеличения селезенки (иногда — печени), можно не обнаружить никаких изменений;
• в период криза симптомы более яркие и представлены повышением температуры тела, более интенсивной желтухой и сосудистыми изменениями по типу синдрома Рейно (особенно при АИГА, провоцируемой действием низких температур).
Информация, полученная на первом и втором этапе, не дает оснований для установления диагноза АИГА, а тем более для идентификации ее серологического варианта. Может возникнуть лишь предположение об этом заболевании (особенно при развитии несомненных гемолитических кризов, синдроме Рейно или выделении черной мочи в период криза). Для уточнения диагноза необходимо доказать аутоиммунность ГА и отвергнуть ряд заболеваний печени и желчных путей, способных сопровождаться сходными симптомами.
На третьем этапе диагностического поиска обнаруживают в большей или меньшей степени выраженный синдром гемолиза (в зависимости от существования или отсутствия гемолитического криза). Чрезвычайно важно обнаружение аутоантител. Основной метод определения неполных тепловых агглютининов — проба Кумбса, основанная на агглютинации антиглобулиновой сывороткой эритроцитов больного с фиксированными на них антителами (прямая проба Кумбса) или агглютинации с помощью антиглобулиновой сыворотки эритроцитов здорового человека, «нагруженных» антителами из сыворотки крови больного (непрямая проба Кумбса). Аутоантитела также обнаруживают с помощью агрегатгемагглютинационной пробы, которая во много раз чувствительнее пробы Кумбса.
Полные холодовые агглютинины обнаруживают путем инкубации при различных температурах эритроцитов донора и сыворотки больного. Агглютинация происходит при определенных разведениях сыворотки и температуре, при этом чем выше температура, при которой возможна агглютинация, тем тяжелее протекает болезнь.
Двухфазные гемолизины определяют с помощью эритроцитов донора, фиксирующих на себе антитела больного при низкой температуре. В дальнейшем при инкубации такой смеси происходит гемолиз эритроцитов. Иногда для обнаружения гемолизинов используют пробу Кумбса: чем более высокая температура требуется для гемолиза, тем тяжелее протекает заболевание.
При формах АИГА, протекающих с выработкой гемолизирующих аутоантител (гемолизинов), в моче присутствуют гемоглобин и гемосидерин, так как гемолиз протекает внутри сосудов. Моча приобретает темную окраску (вплоть до черной).
На третьем этапе диагностического поиска при симптоматических формах АИГА можно обнаружить изменения, обусловленные основным заболеванием:
опухолью, гемобластозом, диффузным заболеванием соединительной ткани, поражением печени и др.
Диагностика АИГА основана на обнаружении сочетания признаков гемолиза и определении аутоантител. Естественно, что в процессе диагностики следует исключить ГА, обусловленные воздействием различных химических средств, малярийного плазмодия, механическим повреждением оболочки эритроцита, а также наследственной этиологии.
При назначении лечения учитывают фазу аутоиммунной ГА (ремиссия или гемолитический криз).
В период криза средством выбора служат глюкокортикоиды, которые всегда прекращают или уменьшают гемолиз. В острой фазе назначают большие дозы преднизолона (по 60-90 мг/сут) или эквивалентные дозы других глюкокортикоидов. При наступлении ремиссии их постепенно уменьшают, переводя больного на поддерживающие дозы (по 5-10 мг/сут). Продолжительность гормонального лечения при проведении гематологического и серологического контроля (до исчезновения или существенного уменьшения количества аутоантител) составляет 2-3 мес.
В межприступном периоде можно назначать другие иммунодепрессанты, например аминохинолиновые препараты (хлорохин), которые следует принимать длительно (до одного года).
При плохой переносимости глюкокортикоидов, противопоказаниях к их применению или недостаточной эффективности рекомендовано применение цитостатических иммунодепрессантов (циклофосфамид, метотрексат). Эти средства особенно эффективны при АИГА, связанной с холодовыми агглютининами.
В случаях, когда применение глюкокортикоидов и цитостатических средств не позволяет достичь четкого улучшения, хороший эффект может оказать спленэктомия. При выраженной анемии рекомендовано переливание эритроцитарной массы, но кровь необходимо подбирать индивидуально, с помощью непрямой пробы Кумбса, когда переливаемые эритроциты «нагружают» антителами сыворотки крови больного. Если проба Кумбса с эритроцитами донора отрицательная, то такую кровь можно переливать.
При незначительном гемолизе и отсутствии гемолитических кризов прогноз удовлетворительный. Усиление гемолиза с резким снижением концентрации гемоглобина значительно ухудшает прогноз.
Меры первичной профилактики ГА в настоящее время разработаны недостаточно. При установлении диагноза ГА больных ставят на диспансерный учет
и периодически проводят исследования крови. Кроме того, им запрещают контакт с веществами, способствующими усилению гемолиза.
Пароксизмальная ночная гемоглобинурия
Пароксизмальная ночная гемоглобинурия (ПНГ), также называемая болезнью Маркиафавы-Микели, — приобретенная ГА, сопровождающаяся постоянным внутрисосудистым гемолизом и выделением с мочой гемосидерина.
В основе заболевания лежит выработка патологического клона эритроцитов с повышенной чувствительностью к различным гемолитическим агентам (тромбин, комплемент, снижение рН крови и др.). Предполагают, что заболевание возникает в результате соматической мутации на уровне клетки-предшественницы миелопоэза, что приводит к выработке патологического клона эритроцитов, оболочка которых дефектна. При сканирующей микроскопии такие клетки выглядят своеобразно — их мембрана имеет множество отверстий. Кроме этой патологической популяции эритроцитов, существуют и нормальные клетки, не разрушающиеся при подкислении среды. Патологические клоны эритроцитов, гранулоцитов и тромбоцитов высокочувствительны к действию комплемента вследствие дефицита в их оболочке регуляторного белка, который частично обеспечивает быстрое превращение активного С3-компонента комплемента в неактивную форму.
При ПНГ гемолиз происходит внутри сосудов, поэтому в крови присутствует свободный гемоглобин. Его обнаружение в моче зависит от содержания гемоглобина в крови и концентрации гаптоглобина — белка крови, связывающего свободный гемоглобин. При достаточном содержании последнего и небольшом гемолизе гемоглобинурии не будет, но при прохождении свободного гемоглобина через канальцы почек он разрушается и откладывается в эпителии канальцев в виде гемосидерина, а также выделяется с мочой (гемосидеринурия), что служит важным признаком болезни. Заболевание протекает волнообразно: периоды умеренно выраженного гемолиза чередуются с его резким усилением — гемолитическими кризами, во время которых концентрация свободного гемоглобина в крови резко увеличивается; его также обнаруживают в моче (гемоглобинурия).
Клиническую картину заболевания определяет выраженность внутрисосудистого гемолиза, а также возникновение множественных тромбозов мелких сосудов (мезентериальных, верхних и нижних конечностей, почечных, мозговых и сосудов селезенки). Предполагают, что склонность к последним, отмечаемая в периоды гемолитических кризов, обусловлена освобождением при гемолизе эритроцитов (особенно ретикулоцитов) и большого количества веществ, обладающих тромбофилической активностью.
На первом этапе диагностического поиска можно обнаружить жалобы больного на периодически возникающую слабость и легкое желтушное окрашивание склер. Типичный признак болезни — моча черного цвета, обусловленная присутствием в ней гемоглобина и гемосидерина. Нередко гемоглобинурия
возникает в ночное время, что объясняется наступающим во время сна физиологическим ацидозом, а также активацией других факторов, усиливающих гемолиз. Во время гемолитических кризов, кроме черной мочи, больные также отмечают приступы болей в животе (капиллярные тромбозы мезентериальных сосудов). В период криза у пациентов может повышаться температура тела. В анамнезе могут быть указания на госпитализацию в инфекционное отделение с подозрением на вирусный гепатит после возникновения желтушности кожного покрова. В отношении части больных такие предположения сохраняются, если присутствует постоянная желтушность, а четко выраженных кризов с отхождением черной мочи нет.
На втором этапе диагностического поиска в период гемолитического криза отмечают бледность кожи с небольшим желтушным оттенком. При развитии достаточно выраженной анемии обнаруживают циркуляторногипоксический синдром (тахикардия, анемический систолический шум, снижение АД, шум «волчка» на яремных венах) и небольшое увеличение селезенки. Возможно увеличение печени, но это не постоянный признак. Данные второго этапа позволяют исключить ряд заболеваний печени, способных вызвать желтуху. В связи с тромбозами сосудов у части больных во время гемолитического криза определяют признаки острого живота (резкие боли, симптомы раздражения брюшины). Нередко это приводит к выполнению хирургических вмешательств, что связано с подозрением на острый аппендицит, прободную язву или острый холецистит.
Решающим в диагностике считают третий этап диагностического поиска, во время которого обнаруживают синдром внутрисосудистого гемолиза.
При общеклиническом исследовании крови обнаруживают снижение концентрации гемоглобина и количества эритроцитов. В период обострения болезни содержание гемоглобина существенно уменьшается (до 30-50 г/л), возвращаясь к норме в период ремиссии. Число эритроцитов уменьшается соответственно снижению концентрации гемоглобина, поэтому цветовой показатель долго остается близким к 1,0. Он снижается, если больной теряет с мочой много железа в виде гемоглобина и гемосидерина. Содержание ретикулоцитов умеренно повышено (до 2-4\%). Количество лейкоцитов чаще всего умеренно снижено, а тромбоцитов — соответствует норме или умеренно снижено.
Исследование костного мозга позволяет обнаружить признаки гемолитической анемии — раздражение красного ростка при нормальном количестве миелокариоцитов.
Концентрация железа в крови снижается вследствие гемоглобинурии, при этом гемосидерин постоянно обнаруживают в моче. Тем не менее снижение содержания железа не считают признаком ПНГ.
Концентрация билирубина в большинстве случаев повышается незначительно или не изменяется.
Нарушение структуры оболочки эритроцита обнаруживают с помощью пробы Хэма (кислотная проба) и сахарозной пробы. При пробе Хэма эритроциты больного гемолизируются в свежей подкисленной сыворотке (нормальные эритроциты не подвержены гемолизу). При сахарной пробе их гемолиз происходит при добавлении к сыворотке сахарозы.
Диагностика заболевания основана преимущественно на данных третьего этапа диагностического поиска (положительный результат кислотного и сахарозного теста), а также на особенностях клинической картины болезни (приступы болей в животе, выделение темной мочи, волнообразное течение болезни). ПНГ необходимо дифференцировать от приобретенной АИГА, при которой в сыворотке крови присутствуют гемолизины, обусловливающие внутрисосудистый гемолиз. Последний также манифестирует гемоглобинемией, гемоглобинурией и гемосидеринурией. Дифференциальная диагностика также основана на обнаружении гемолизинов, для чего выполняют пробу Кумбса.
Патогенетических методов лечения ПНГ не существует. Тяжесть анемии обусловлена интенсивностью гемолиза и ответной реакцией клеток эритроидного ряда. При анемии целесообразно переливание эритроцитов, предварительно трижды отмытых изотоническим раствором хлорида натрия. Такие эритроциты переливают 1 раз в 4-5 дней в дозе 200-400 мл. Многие больные нуждаются в таких переливаниях на протяжении нескольких месяцев.
В последнее время применяют средства, обладающие антиоксидантным действием и способствующие стабилизации мембран эритроцитов. Витамин Е назначают в дозе 3-4 мг/сут.
Препараты железа назначают при его значительной потере или выраженном дефиците.
Для борьбы с тромбозами, как правило, в небольших дозах применяют гепарин натрия (по 5000 ЕД 2-3 раза в день под кожу живота), а также антикоагулянты непрямого действия.
Продолжительность жизни пациентов составляет от одного года до семи лет с момента возникновения первых признаков заболевания. Кроме того, описаны случаи значительной продолжительности жизни больных. Редко регистрируют полную ремиссию и даже полное выздоровление.
источник