Н. Р. Аблаев, д. м. н., профессор, А. С. Искакова, А. Ж. Дуйсенбаева,
кафедра лабораторной диагностики и молекулярной медицины
КазНМУ им. С. Д. Асфендиярова, г. Алматы
Еще больше неточностей и просто заблуждений относительно анемии, особенно т. н. железодефицитной анемии (ЖДА) в современной медицине.
Начало XXI века ознаменовалось открытием нового гормона, регулирующего метаболизм железа в организме человека. Это гепсидин. Железо – незаменимый элемент для роста и выживания организмов, играет важную роль в многочисленных биологических функциях. Его участие особенно очевидно в транспорте кислорода гемоглобином, в синтезе ДНК (в составе коэнзима редуктазы рибонуклеотидов) и в активности оксидоредукции многочисленных митохондриальных энзимов.
Количество железа в организме чрезвычайно стабильно и определяется равновесием между поступлением и потерей этого металла.
В организме взрослого человека содержится приблизительно 4–4,5 г железа. Это обеспечивается за счет очень небольшой части поступающего с пищей и в основном за счет рециклирования железа, начиная с лизиса старых эритроцитов. В этом последнем механизме особенно задействованы макрофаги селезенки и красного костного мозга, и в меньшей мере клетки Kupffer. От 60 до 70% железа входит в состав гемоглобина. Приблизительно 10% находится в миоглобине, цитохромах и ферментах, содержащих железо. Остальное железо переходит в запас железа или в форме ферритина (легко мобилизируемая форма резерва), или в форме гемосидерина (трудно мобилизуемая форма резерва). Плазматический транспорт включает трансферритиновое железо и составляет приблизительно 1% железа от общего объема организма.
Ежедневные потери железа – 1–2 мг. В основном они осуществляются через пищеварительный тракт: в виде десквамации эпителиальных клеток кишечника, микрокровотечений и потери с желчью.
Железо также теряется и при десквамации эпителиальных клеток кожи, и в меньшей степени с мочой. Компенсация этих потерь происходит путем абсорбции железа из пищи. Регуляция этой абсорбции находится под воздействием общего содержания железа в организме, эритропоэтической активности, гипоксии и содержания железа в пище и природы питания, а в последнее время очень важное значение в данном процессе придается гормону гепсидину.
Энтероциты ворсинок двенадцатиперстной кишки и проксимальной части jejunum ответственны почти за полную абсорбцию геминического и негеминического железа. Эти энтероциты являются результатом созревания и миграции мультипотентных исходных клеток, располагающиеся в дуоденальных криптах. Чтобы попасть из интестинального просвета в плазму, железо должно пересечь апикальную мембрану, сам энтероцит, а затем базолатеральную мембрану, т. е. путь железа не простой.
Геминическое железо эндоцитируется с молекулой гема после сливания с потенциальным рецептором, называемым гемпереносящим белком (HTP). Затем железо освобождается в энтероците после отторжения молекулы гема гем-оксигеназой.
Что касается абсорбции железа не геминического (неорганического, окисленного) на уровне апикального полюса энтероцита, то наиболее вероятный механизм – это участие белка-транспортера дивалентного катиона DMT1. В этом случае атомы феррического железа (окисленного, трехвалентного железа), поступившие с пищей, сначала редуцируются в атомы двухвалентного железа ферриредуктазой, называемой Cybrd1, затем захватываются транспортером DMT1 (трехвалентные атомы металлов через этот канал пройти не способны).
Из сказанного и показанного на рисунке 1 видно, что основной причиной анемии является не отсутствие необходимого количества железа в пище, а дефекты того или иного белковой природы соединения на долгом и трудном пути усвоения и использования атомов железа. Возможно, будут открыты и другие пути и механизмы развития анемии на фоне нормального поступления железа с продуктами питания.
Потенциальная токсичность железа (восстановленное железо превращает часть молекулярного кислорода в супероксидный радикал) предполагает, что он находится в комплексе с мелкими молекулами или внутриклеточными протеинами шаперонами. После захвата железо может быть направлено в сторону ферритина, где протеины энтероцита могут использовать его в качестве неорганического кофактора, или к базальному полюсу энтероцита. В первом случае абсорбированное железо будет утрачено в процессе естественной десквамации энтероцитов, тогда как во втором оно будет находиться в резерве, чтобы быть направленным в общую циркуляцию. Процесс высвобождения железа в сторону общей циркуляции включает, по меньшей мере, два протеина, которые были идентифицированы совсем недавно.
Ферропортин экспортирует железо из энтероцитов в кровь. Присутствует в макрофагах, 12-перстной кишке, печени и плаценте. Он встречается только у позвоночных. По последним данным, ферропортин служит также рецептором для гепсидина. После прикрепления гепсидина – гормона – к ферропортину весь комплекс поступает в цитоплазму энтероцита и разрушается, и прежде всего белок-канал ферропортин, а возможно, и гепсидин. В результате базолатеральная мембрана лишается канала для выхода железа в сторону крови (обычная мембрана клеток вообще не пропускает катионы железа ни в ту, ни в другую сторону).
При отсутствии гепсидина или при незначительных его (базальных) количествах Fe2 + выходит из энтероцита и тут же под влиянием фермента гефестина (феррооксидазы) окисляется до Fe3 + , которое подхватывается трансферрином (одна молекула трансферрина связывает два атома окисленного железа). Гепсидин синтезируется в печени. Молекула активного гормона состоит из 25 аминокислотных остатков и содержит 4 дисульфидных связей (рис. 4). Гепсидин блокирует поступление железа в плазму. Блокирует экспорт железа из макрофагов, содержащих Fe отмирающих эритроцитов. Таким образом, гепсидин связывается с ферропортином и индуцирует его интернализацию (перемещение внутрь клетки) и далее деградацию. С помощью такого механизма гепсидин регулирует выход железа из клеток 12-перстной кишки в плазму и тем самым обусловливает распределение железа в организме. Когда гепсидина в крови много (воспаление), он вызывает анемию, когда его мало, конечно, при наличии нормальных ферропортинов, развивается гемохроматоз (избыточное поступление Fe из кишечника и депонирование железа во многих тканях).
Гепсидин блокирует поступление железа в плазму. Блокирует экспорт железа из макрофагов, содержащих Fe отмирающих эритроцитов. Таким образом, гепсидин связывается с ферропортином и индуцирует его интернализацию (перемещение внутрь клетки) и далее деградацию. С помощью такого механизма гепсидин регулирует выход железа из клеток 12-перстной кишки в плазму и тем самым обусловливает распределение железа в организме. Когда гепсидина в крови много (воспаление), он вызывает анемию, когда его мало, конечно, при наличии нормальных ферропортинов, развивается гемохроматоз (избыточное поступление Fe из кишечника и депонирование железа во многих тканях).
Название гепсидин происходит из следующих компонентов: «геп» – от hepar (печень), окончание «сидин» указывает на его антибактериальные свойства.
Первоначально (2000 г.) гепсидин считался одним из новых белков острой фазы воспаления. Но через год было обнаружено, что он синтезируется в печени и по всем параметрам должен быть отнесен к гормонам. Таковым он сейчас и рассматривается.
Дисрегуляция гепсидина приводит к ряду патологий. При анемии воспаления усиливается секреция гепсидина печенью, гепсидин-секретирующими опухолями; продукция гепсидина при этом является очень высокой, а концентрация железа в плазме становится очень низкой, что неминуемо ведет в анемии. При врожденном гемохроматозе и железо-обусловленных анемиях продукция гепсидина неадекватно низкая, поэтому абсорбция и высвобождение железа из соответствующих клеток очень высокие. Концентрация железа в крови также очень высокая. Это приводит к перенасыщению тканей железом, следовательно, в таких тканях налицо железотоксичность (гемохроматоз).
Вполне возможно, появятся (уже появляются) данные, говорящие о том, что гепсидин имеет не только печеночное происхождение, но производится также рядом других органов и тканей (желудке, сердце). Такова научная история многих открытий: гормональное соединение, выделенное из гипоталамуса или из другой железы, через некоторое время обнаруживается в достаточно эффективных количествах в поджелудочной железе, в клетках, разбросанных в различных отделах ЖКТ.
Механизм развития анемии при хронических воспалительных заболеваниях выглядит следующим образом: в ответ на липополисахарид бактериальной стенки (ЛПС), а также на секретируемые фагоцитами провоспалительные интерлейкины (ИЛ-1, ИЛ-6), гамма-интерферон, TNF-α происходит притупление эритропоэтинового ответа, снижение ответа эритроидных тканей на ЭПО, уменьшается поставка железа из РЭС, повышается содержание ферритина (железа) внутри клеток, уменьшается уровень железа в трансферрине и, следовательно, повышается коэффициент насыщения трансферрина, снижается абсорбция железа из ЖКТ. Причинами указанных явлений служат:
– воспаление, некроз тканей, инфекции;
– неоплазия;
– различная патология сердечно-сосудистой системы, инфаркт миокарда и др.
Относительно недавно стало известно, что анемия развивается также при дефиците витамина D: недостаточное содержание витамина D в организме не позволяет кальцитриолу (активный метаболит витамина D3) стимулировать ген, кодирующий белок-рецептор эритропоэтина в красном костном мозге: эритропоэтин (ЭПО), синтезирующийся почках, может регулировать эритропоэз только через свои рецепторы. При достаточном уровне кальцитриола, гормонально активного метаболита витамина D, происходит стимуляция гена, кодирующего белковой природы рецепторы ЭПО, которые далее запускают синтез в клетках красного костного мозга гемоглобина и эритроцитов. При дефиците витамина D эти процессы становятся невозможными. В мире насчитывается более 2 млрд человек с ЖДА. Какая часть из них страдает именно из-за дефицита витамина D, остается большой тайной. По сообщениям мирового эксперта по витамину D М. Холика, в целом до 70–80% человечества имеют явный дефицит данного солнечного витамина. Но всех больных с ЖДА без особых успехов лечат препаратами железа, а не витамином D.
Вполне возможно, со временем откроется взаимосвязь между дефицитом витамина D и усиленной секрецией гепсидина. Уже считается доказанным факт, что кальцитриол обладает выраженными противовоспалительными свойствами. А как говорилось выше, гепсидин начинает проявлять свое действие именно в ответ на воспаление. Так что применение витамина D при ЖДА окажется весьма успешным и многозначительным.
На схеме (рис. 6), состоящей из нескольких фрагментов, показано, как в норме происходит усвоение и использование атомов железа для гемопоэза (1), а также как гепсидин при воспалительных процессах высвобождается из печени и блокирует выход железа из дуоденальных энтероцитов, макрофагов и гепатоцитов, приводя неминуемо к дефициту железа в крови, хотя больные при этом могут получать вполне нормальные количества железа с пищей (2). На фрагменте 3 представлены случаи, когда имеет место гепсидинорезистентность вследствие дефекта его рецептора, роль которого выполняет ферропортин: железо при этом беспрепятственно наводняет кровь, откладывается во многих органах и тканях, способствуя развитию гемохроматоза 2-го типа.
Избыточное поступление железа в органы и ткани вызывает гемохроматоз. Большинство форм врожденного гемохроматоза являются следствием генетического дефекта гепсидин-регулирующих белков HFE, рецептора трансферрина-2, гемоювелина или самого гепсидина. При всех указанных случаях продукция гепсидина становится очень низкой, и потеря контроля со стороны гепсидина приводит к беспрерывному всасыванию железа из продуктов питания, насыщению транс-феррина плазмы, токсически высокому отложению железа в печени и других органах.
Железо, поступающее из энтероцитов (5%) и из мононуклеарных макрофагов (95%), в нормальных условиях в основном переводится в костный мозг, где оно необходимо для синтеза гемоглобина. Фракция железа, не предназначенная для костного мозга, делится между другими различными местами утилизации и местами накопления представленными макрофагами, но в основном, гепатоцитами, особенно чувствительными к перегрузкам железом. Примеры развития анемии воспаления представлены на рисунке 7.
Железо транспортируется в плазму в основном в форме железа связанного с трансферрином. Комплекс железо–трансферрин затем захватывается рецептором-1 трансферрина (RTf1), присутствующим в различных органах, в частности печени и эритропоэтических клетках. Во время перегрузок железа появляется особая биохимическая форма железа. Речь идет о железе, не связанном с трансферрином, особенностью которого, в отличие от железа, связанного с трансферрином, является преимущество захвата печенью. Эта форма железа, способного генерировать свободные радикалы, не поддается любой из известных форм регуляции и в весьма значительной степени способствует осложнениям, связанным с перегрузками железом.
Высокий уровень железа и снижение уровня кислорода тормозят секрецию гепсидина. Воспалительные (хронические) заболевания стимулируют секрецию фагоцитами провоспалительных цитокинов, которые вызывают синтез и секрецию печенью гормона гепсидина (он же один из белков острой фазы воспаления) и снижение синтеза эритропоэтина. Эритропоэтин должен запустить процесс формирования клеток крови из плюрипотентных стволовых клеток. Поскольку гепсидин при этом разными путями уменьшает уровень железа в крови (путем снижения абсорбции, транспорта и биодоступности) и трансферрин не доставляет железо в костный мозг, то включение атома железа в протопорфирин IX не происходит, не сформировавшиеся клетки крови подвергаются апоптозу. В результате развивается анемия.
Гепсидин как лабораторный тест интересен еще и с других точек зрения: во-первых, мутации и другие воздействия на печеночные гены, кодирующие гепсидин (это, как правило, врожденные дефекты), разрушение самих гепатоцитов приводят к неспособности печени синтезировать и секретировать нужное количество гепсидина, – тогда нарушается возможность регуляции ферремии, что неминуемо приводит к избыточному отложению железа в различных органах и тканях (рис. 9),  вследствие чего развивается гемохроматоз 1-го типа (ювенильный гемохроматоз; во-вторых ген, кодирующий ферропортин, оказывается, тоже может стать дефектным, т. е. неспособным взаимодействовать с гормоном гепсидином, выход железа из соответствующих клеток не контролируется, развивается гиперферремия с развитием гемохроматоза 2-го типа.
вследствие чего развивается гемохроматоз 1-го типа (ювенильный гемохроматоз; во-вторых ген, кодирующий ферропортин, оказывается, тоже может стать дефектным, т. е. неспособным взаимодействовать с гормоном гепсидином, выход железа из соответствующих клеток не контролируется, развивается гиперферремия с развитием гемохроматоза 2-го типа.
Если гемохроматоз 1-го типа является гепсидин-зависимым и может быть излечен путем введения экзогенного гепсидина (пока гепсидин как лекарственное средство не существует, но скоро должен стать доступным), то для лечения гемохроматоза 2-го типа (гепсидин-независимый тип) должны подбираться другие методы терапии.
Имеются основания предполагать, гемохроматозы чаще всего не диагностируются. Повсеместное внедрение в лабораторную практику исследования гепсидина (прогепсидина), безусловно, поможет исправить положение.
Гемохроматоз вызывается мутациями в гене HFE. Исследователи идентифицировали более 20 мутаций в гене HFE, которые вызывают гемохроматоз 1-го типа. Эти мутации изменяют размер белка или его трехмерную структуру. В результате белок не может функционировать правильно. Характерна замена аминокислоты цистеина на тирозин в позиции 282 в белковой цепи (описывается как C282Y или Cys282Tyr). Другая мутация вызывает замену гистидина на аспарагиновую кислоту в положении 63 (описывается как H63D или His63Asp). В результате таких замен измененный белок не удерживается на поверхности клеток и не взаимодействует с рецептором на клеточной поверхности, рецептором для трансферрина. Рецептор трансферрина играет ключевую роль в регулировании количества железа, которое проникает в клетки. Когда же белок HFE не связывается с рецептором трансферрина, слишком много железа поступает в организм, проходя бесконтрольно через стенку тонкого кишечника. Такая чрезмерная абсорбция железа (перегрузка организма железом) обусловливает характеристики данного заболевания порфирия – повышенный риск вследствие мутаций в гене HFE.
Mутации в гене HFE, которые вызывают гемохроматоз, обусловливают также особую форму порфирии, называемую porphyria cutanea tarda.
Цитогенетическая локация гена HFE: 6p21.3
Молекулярная локация хромосомы 6: пары оснований 26,195,426 to 26,205,037
Представление о молекулярных механизмах контроля гомеостаза железа быстро углубляется за последние годы благодаря методам молекулярной генетики и идентификации большого количества протеинов, участвующих в метаболизме этого металла. Анализ экспериментальных моделей (носителей естественных мутаций или экспериментальных) и изучение заболеваний человека, связанных с нарушениями метаболизма железа, также внесли свою лепту. Они позволили выявить комплексность биохимических процессов, задействованных в поддержании баланса железа в организме, хотя и без достаточного понимания всех механизмов. Имеются многочисленные аргументы в пользу того, что интестинальная гиперабсорбция железа не является единственной причиной генетического гемохроматоза, но что макрофаги, как и клетки печени, по меньшей мере, играют такую же важную роль, как и энтероциты в развитии этой наследственной перегрузки железом.
Традиционная система контроля ЖДА органами ВОЗ заставляет лечащих врачей даже на основе однократного исследования уровня гемоглобина и обнаружения при этом еще совсем неясной природы анемии назначать больным препараты железа. Но, как показывает практика, ферротерапия дает положительный эффект лишь в половине случаем, а на остальную половину больных с «ЖДА» препараты железа оказывают отрицательный эффект, вплоть до генерализации воспалительного процесса. Настало время точно выяснять, что же у конкретного больного – ЖДА или АХЗ, почему и как они развились. Если больной страдает анемией воспаления (об этом опытный врач подумает, если в общем анализе крови, крови гипогемоглобинемии, отмечаются повышенная СОЭ, лейкоцитоз, повышенный уровень СРБ, т. е. маркеров воспаления), то необходимо вначале убрать воспаление. При таком подходе повторные анализы крови укажут на динамику гемоглобинемии, а возможно, и на нормализацию уровня гемоглобина. Больной, таким образом, будет избавлен от негативного действия продолжительной железотерапии.
Новый этап исследований этого метаболизма состоит в дальнейшем изучении экспрессии совокупности генов различных типов клеток с помощью нанотехнологий в различных условиях перегрузки или недостатка железа.
Разумеется, приведенными здесь фактами не ограничивается сущность всех серьезных «железных» проблем. Читатель по ходу знакомства с данным материалом, наверное, обратил внимание на большую группу белков, задействованных в метаболизме железа. Если учесть, что эти белки кодируются генами, которые, к сожалению, обладают способностью подвергаться различным мутациям и производить дефектные белки, то становится понятным существование множества форм анемий, передающихся от поколения к новым поколениям и не поддающихся лечению только приемом железа.
К сказанному следует также добавить, что содержание гепсидина в крови определятся с помощью метода ИФА. Изучение изменений данного показателя, совместно с исследованиями разных параметров эритроцитов, уровня гемоглобина в крови, а также ряда белков острой фазы воспаления внесет существенный вклад в развитие гематологии, заставит клиницистов учитывать возможности развития анемии у больных с хроническими заболеваниями, которые к тому же сопровождаются оксидативным стрессом.
У здоровых добровольцев, по данным Tomas Ganz, Gordana Olbina, Domenico Girelli, Elizabeta Nemeth and Mark Westerman (Blood, 15 November 2008, Vol. 112, No. 10, pp. 4292–4297), концентрация гепсидина в крови у мужчин колеблется в пределах 29–254 ng/mL (n=65), а у женщин несколько ниже – 17–286 ng/mL (n=49), (р 10 mg/ dL), миеломой, хронической почечной патологией, наоборот, концентрация гепсидина была очень высокой. По мнению авторов, измерение уровня гепсидина в крови является очень перспективным маркером, особенно при затруднениях отделения физиологических изменений от патологических нарушений, а также для выявления генетических дефектов, в том числе в случаях ювенильного гемохроматоза, уточнения этиологии заболеваний, обусловленных нарушением метаболизма железа. Но, конечно, можно себе представить картину, какое появится противодействие со стороны производителей железных препаратов, когда, в результате правильной диагностики анемий и этиопатогенетической терапии, железо будет все больше ржаветь на складах аптек. Через это человечество когда-нибудь научится проходить.
– Гемоглобин – действительно единственный переносчик кислорода в организме.
– При снижении уровня гемоглобина падает не только снабжение тканей кислородом, но также снижается эффективность борьбы с закислением крови и выведение углекислого газа из организма.
– Не всегда нормальные цифры общего гемоглобина означают благополучие с доставкой кислорода к органам и тканям.
– При обнаружении сниженного гемоглобина в анализах у больного не спешите кричать, что у него ЖДА, и немедленно приступать к назначению ему препаратов железа и только железа.
– Медицина, как и сама жизнь, – это сложная штука; истина, как правило, не лежит на поверхности разных протоколов и стандартов, ее необходимо хорошенько поискать. И вы обязательно найдете причину!
источник
Анемия хронических заболеваний (ее по-другому еще называют анемией воспаления) является распространенным типом патологии, которая развивается у больных, страдающих тем или иным инфекционным, воспалительным или опухолевым заболеванием. Отличительной особенностью такой анемии является понижение сывороточного железа, но, в отличие от истинной нехватки железа, этот микроэлемент может сохраняться в макрофагах.
Анемия хронических заболеваний выступает в настоящее время наиболее распространенной проблемой. Этот тип недуга занимает второе место после железодефицитной анемии. Данная патология может сопровождать любой инфекционный, ревматический или опухолевый недуг, а, кроме того, недостаточность сердца, хронические болезни почек, диабет, цирроз печени и так далее.
Анемию хронических заболеваний (АХЗ) определяют при инфекционном процессе, который ассоциирован с микробными патогенами (бактериальными, вирусными или грибковыми инфекциями), а кроме того, с аутоиммунными заболеваниями, в частности, с такими как системная волчанка, ревматоидный артрит и другие. К возникновению анемии хронических патологий также ведут хронические заболевания, которые сопровождаются воспалением низкой градации, например, онкологическое новообразование, хронические болезни почек, недостаточность сердца и тому подобное. Кроме того, отмечается подобный патогенез анемии хронических заболеваний в ходе старения, на фоне чего у больных отмечают активацию воспалительных цитокинов.
Исследования, которые были проведены в последние десятилетия, позволяют установить патофизиологический механизм анемии хронических заболеваний. Болезни, которые сопровождаются дефицитом железа, весьма многочисленны. Но главным является железодефицитная недостаточность наряду с АХЗ.
Сложность для медиков представляет в первую очередь дифференциальная диагностика анемий хронических заболеваний. При наличии анемии хронических патологий имеет место гипохромный дефицит гемоглобина с низким показателем сывороточного железа, но с увеличенным при этом ферритином. Стоит отметить, что лечение такой анемии с помощью препаратов железа не ведет к компенсации эритропоэза. Использование современных диагностических исследований позволяет улучшать и ускорять диагностику анемии.
Учитывая то, что анемия хронических заболеваний выступает вторичным проявлением базового заболевания, терапия последнего корригирует и анемию. Правда, такая терапия не всегда возможна. Современной тенденцией в медицине является изучение молекул новых лекарственных средств, чьей мишенью служат основные патогенетические звенья хронических заболеваний, в особенности это цитокины наряду с корректорами ветви ферропортина. Но большая часть лекарств находится еще на стадии экспериментального исследования.
Картина крови при описываемом недуге наблюдается следующая:
- Показатель сывороточного железа снижен.
- При наличии у пациента анемии хронических заболеваний железосвязывающая способность эритроцитов будет понижена. Если же этот показатель увеличен, то можно исключить дефицит гемоглобина. Правда, изменение данного значения не является специфичным признаком для того, чтобы дифференцировать анемию хронических недугов от железодефицитной болезни.
- При данном диагнозе насыщенность сыворотки трансферрином, как правило, бывает в норме. Значение выше десяти процентов говорит о снижении железа. А показатель менее десяти процентов свидетельствует о наличии дефицита этого микроэлемента. Железодефицитная болезнь может быть во многом связана с желудочно-кишечными кровотечениями вследствие лечения рефрактерной анемии посредством нестероидных противовоспалительных препаратов.
- На фоне этого заболевания сывороточный ферритин обычно бывает в норме или повышается в противоположность недостаточности железа.
- При наличии ревматоидного артрита, болезней печени или на фоне новообразований нормальное значение сывороточного ферритина не исключает сопутствующий дефицит железа. Правда, уровень ферритина менее 40 нанограмм на миллилитр указывает на значительное снижение запасов железа в организме.
- Такой показатель, как свободный эритроцитарный порфирин, при наличии анемии хронических патологий будет повышен.
Анемия хронических патологий по причине своего медленного развития и протекания в легкой форме, как правило, практически никакой симптоматики не дает. Любые проявления имеют отношение обычно к тем недугам, на фоне которых либо вследствие которых в организме возникает анемия.
Итак, к симптомам, которые свойственны развивающейся анемии, можно отнести наличие у пациентов повышенной утомляемости организма наряду с общей его слабостью и резким снижением работоспособности. Помимо всего прочего, к характерной симптоматике стоит причислить явную раздражительность с частыми головокружениями, сонливостью, шумовыми ощущениями в ушах, мушками перед глазами, учащенным сердцебиением и одышкой при физической нагрузке или даже в состоянии покоя.
Таким образом, в случае возникновения подобной симптоматики стоит начинать бить тревогу и обращаться к врачу для проведения необходимых диагностических исследований и дальнейшего адекватного лечения.
Лучше выяснить заранее, что это такое анемия и чем опасна данная патология.
Анемии свойственны определенные общие признаки. Обычно это наличие нормоцитарной анемии легкой степени тяжести, когда гемоглобин держится в районе более 90 грамм на литр. Такая анемия развивается в течение первых двух месяцев при наличии инфекций, воспалительной патологии либо на фоне злокачественного образования, при этом она не прогрессирует. При показателе гемоглобина ниже 80 грамм на литр следует подумать о присутствии дополнительных факторов, которые участвуют в патогенезе анемии. Кроме этого, выраженность анемии зачастую может коррелировать с продолжительностью и активностью базового заболевания (хроническая инфекция, болезни соединительных тканей и так далее).
Все методы, применяемые для диагностики анемии хронических заболеваний, напрямую зависят от самого основного недуга, на фоне которого развивается дефицит железа в организме. Но, тем не менее, в том случае, если анемия имеет место, то в обязательном порядке больным назначается сдача общего и биохимического анализа крови наряду с пункцией костного мозга для того, чтобы установить характер и тип анемии.
Помимо всего прочего, во время диагностики требуется исключить такие причины нехватки железа, как наличие травматических кровотечений и внутренних кровопотерь.
При сборе у пациентов жалоб, как правило, выясняют наличие у больного следующих симптомов:
- Сердцебиения и одышки, усиливающейся при физической нагрузке.
- Головокружений и шума в ушах.
- Слабости и повышенной утомляемости.
Как лечится анемия хронического заболевания (по МКБ-10, кстати, код недуга — D63.8)?
Учитывая то, что анемия хронических заболеваний выступает вторичным проявлением базового заболевания, терапия последнего будет корригировать и дефицит железа. Однако, такая терапия не всегда возможна. К принципам ведения больных с анемией хронических патологий относят следующие пункты:
- Проведение лечения основного заболевания.
- Использование специфических средств лечения анемии. Таковые назначаются только при наличии тяжелой степени болезни, которая ограничивает трудоспособность наряду с повседневной активностью пациента.
- При развитии тяжелой анемии назначается переливание эритроцитарных масс.
- Назначение эритропоэзстимулирующих препаратов с сочетанием с внутривенными медикаментами железа.
- Методики лечения дополнительно могут включать в себя различные эритропоэзстимулирующие инновационные средства наряду с антицитокиновыми препаратами и лекарствами, влияющими на гепсидин и ферропортин.
Стоит отметить, что патология не выступает зарегистрированным показанием к назначению пациентам эритропоэзстимулирующих лекарств, однако таковые могут быть рассмотрены в качестве альтернативного лечения для замещения многократных трансфузий эритроцитов. В некоторых исследованиях отмечаются положительные результаты использования эритропоэзстимулирующих средств в лечении анемии хронических заболеваний.
Среди пациентов с хронической недостаточностью сердца распространенность анемии составляет тридцать семь процентов. Среди этого числа более чем у половины пациентов регистрируют анемию хронических заболеваний. В целом, общая распространенность железодефицитной болезни у пациентов с недостаточностью сердца колеблется от четырнадцати до пятидесяти шести процентов. Такой широкий диапазон напрямую связан с отсутствием единого утвержденного подхода к диагностике анемий, а, кроме того, с возрастными различиями больных.
В настоящее время доказано, что у страдающих недостаточностью сердца пациентов чаще выявляют нормоцитарную анемию, которая составляет до пятидесяти семи процентов случаев. Чаще всего это заболевание бывает ассоциировано с дисфункцией почек и понижением секреции эритропоэтина. Для хронического течения заболевания свойственна плохая утилизация железа наряду с выраженной активацией цитокинов, что на сегодняшний день встречается у пятидесяти трех процентов пациентов.
Возникновению анемии при хронической сердечной недостаточности способствует, как правило, возникновение в крови дефицита железа. В настоящее время доказано, что основными причинами появления анемии у пациентов с недостаточностью сердца являются гемодилюция наряду с хроническими заболеваниями почек и дефицитом витамина В12.
К таковым можно отнести все методики профилактики хронических недугов и их рецидивов. Одной из рекомендаций является соблюдение правильного и сбалансированного питания, богатого железом. Таким образом, для профилактики любых анемий врачи рекомендуют делать упор на мясные и рыбные блюда, так как именно в них содержится наибольшее число этого столь необходимого для организма микроэлемента. В этой связи необходимо отметить, что в особенности больше всего железа содержится в красных сортах мяса, например в говядине. Не стоит забывать и о фруктах, например, свое предпочтение следует отдавать яблокам, гранатам и так далее.
Всем известно, что движение наряду с прогулками являются отличной профилактической мерой при наличии любого заболевания. В связи с этим для предупреждения симптоматики неприятной анемии крайне важно регулярно поддерживать свой организм в тонусе. Занятия умеренными физическими нагрузками в виде фитнеса, аэробики, плавания и лыж, значительно улучшают кровообращение, способствуя в целом хорошему самочувствию.
Помимо всего прочего, нельзя забывать о том, что любая анемия в первую очередь является недостатком кислорода. Поэтому самой лучшей профилактикой анемии считается возможность пополнения запасов кислорода в организме. Для этого врачи советуют много гулять на свежем воздухе. К сожалению, в настоящее время у большинства людей сидячая работа, многие постоянно находятся в душном помещении, а это все непременно сказывается на здоровье организма и отражается на нем не лучшим образом.
Все вышеперечисленные рекомендации являются довольно действенными при наличии дефицита железа любой разновидности и при анемии хронических заболеваний в том числе. Главное, что все эти рекомендации достаточно просты, и следовать им может абсолютно каждый человек. Можно, разумеется, периодически употреблять для профилактики и железосодержащие препараты, но сразу стоит отметить, что злоупотреблять таковыми все же не стоит. Поэтому лучше всего предупредить заболевание, которое может вызвать дефицит железа в организме посредством банального ведения здорового образа жизни.
Мы выяснили, что это такое анемия и чем опасна данная болезнь.
источник
Ф.Ю.Копылов, Д.Ю.Щекочихин
Первый МГМУ им. И.М. Сеченова Кафедра профилактической и неотложной кардиологии ФППОВ, Москва
Анемия с давних времен является спутником человечества. Выдающийся немецкий врач Йоханнес Ланге (Johannes Lange) уже в 1554 г. дал название анемии как «болезни девственниц (morbus virgineus)». Он считал эту болезнь специфичной для целомудренных девушек, а причиной указывал задержку менструальной крови [1], ссылаясь на описание Гиппократа, представленное в сочинении «О болезнях девушек».
За последующие несколько столетий мы существенно продвинулись в понимании патофизиологии анемии, и в настоящий момент крупные исследования посвящаются оценки роли анемии в развитии и течении различных заболеваний на популяционном уровне. Касательно сердечно-сосудистых заболеваний (ССЗ) доказано, что анемия является независимым фактором риска неблагоприятных исходов ССЗ у пациентов с хронической сердечной недостаточностью (ХСН) и хронической почечной недостаточностью (ХПН). Для общей популяции больных ССЗ большинство данных свидетельствует о том, что анемия является и здесь независимым фактором риска, однако данных для включения в официальные рекомендации пока недостаточно [2].
Определение понятия и распространенность
Согласно определению ВОЗ, анемия регистрируется у взрослых женщин при снижении концентрации гемоглобина ниже 12 г/дл, а у мужчин ниже 13 г/дл [3]. На рис. 1 проиллюстрировано применение данных критериев на примере крупной выборки исследования NHANES III (n=40 000) [4]. Применение критерия 12 г/дл для женщин позволяет включить в число страдающих анемией существенно большее число лиц, по сравнению с мужчинами. Этот факт находит свое отражение и в других исследованиях, в которых распространенность анемии у женщин почти в 3 раза превышает таковую у мужчин: 13 и 4,8% соответственно [5], и должен быть принят во внимание при оценке результатов различных исследований.
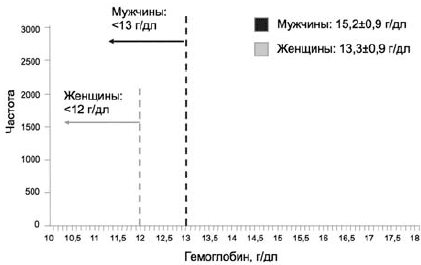
Рис 1. Применение критериев ВОЗ для определения анемии на популяционном уровне [4]
Распространенность анемии среди пациентов с сердечно-сосудистыми заболеваниями относительно хорошо изучена. Среди пациентов с ишемической болезни сердца (ИБС) составляет от 10 до 30% [6, 7], а на примере ХСН — варьирует в широком диапазоне от 4 до 61% (в среднем 18%) в зависимости от тяжести основного заболевания и применяемых критериев анемии [8].
Этиология и патогенез
Причинами анемии у пациента кардиологического профиля могут быть все факторы характерные для общей популяции. Однако если рассматривать группу больных с прогрессирующими ССЗ и в первую очередь с ХСН, то основными этиопатогенетическими факторами анемии можно считать следующие:
1. Анемия хронических заболеваний.
2. Анемия за счет гемодилюции (псевдоанемия).
3. Анемия за счет недостатка железа/витаминов.
4. Нарушение функции почек — снижение продукции эритропоэтина.
5. Действие лекарств.
Анемия хронических заболеваний
Анемия, возникающая у пациентов с инфекцией, воспалением, неоплазиями, хронической почечной недостаточностью и продолжающаяся более 1-2 мес, обозначается как анемия хронических заболеваний (АХЗ) — «анемия воспаления», «цитокинопосредованная анемия». Характерной чертой этого типа анемии является сочетание пониженного уровня железа сыворотки с достаточными его запасами в ретикулоэндотелиальной системе (РЭС). АХЗ по распространенности занимает 2-е место среди анемий (после железодефицитной — ЖДА) [9]. В случае наличия ХСН данный вид анемии является самым распространенным и отмечается у 58% пациентов [10].
В настоящее время считается, что в основе АХЗ лежит иммуноопосредованный механизм: цитокины и клетки РЭС вызывают изменения в гомеостазе железа, пролиферации эритроидных предшественников, продукции эритропоэтина и продолжительности жизни эритроцитов [11]. Открытие гепсидина (hepcidin) — железорегулирующего острофазового белка — позволило во многом прояснить связь между иммунным механизмом нарушения гомеостаза железа и развитием АХЗ: именно через усиление синтеза в печени гепсидина под влиянием воспалительных стимулов, (главным образом интерлейкина-6), происходят снижение абсорбции железа в кишечнике и блокирование высвобождения железа из макрофагов (рис. 2). Дизрегуляция гомеостаза железа ведет к последующей недостаточности доступного для эритроидных предшественников железа, ослаблению пролиферации этих клеток вследствие негативного влияния на них нарушения биосинтеза гема.
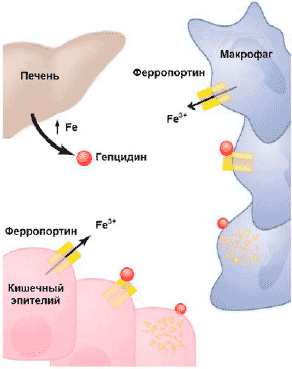
Рис 2. Механизм действия гепцидина: угнетение всасывание железа в кишечнике, замедление мобилизации железа из депо, способствуя его накоплению в макрофагах
Анемия вследствие гемодилюции (псевдоанемия)
Данная причина анемии связана с избыточным «разбавлением» крови и характерна для пациентов с повышенным объемом плазмы (ХСН, ХПН, беременность). Предполагается, что у многих больных ХСН анемия может быть вызвана гемодилюцией [12]. Однако, несмотря на увеличение общего объема плазмы у всех пациентов с систолической и у 71% с диастолической ХСН, истинный дефицит эритроцитов имеется у 88% больных анемией при диастолической ХСН и у 59% при систолической ХСН [13].
Анемия вследствие недостатка железа/витаминов
Еще 50 лет назад было показано непосредственное влияние железодефицита на ферментативные процессы, даже в отсутствии анемии [14]. Экспериментальные исследования на животных показали возможность непосредственного влияния железодефицита на диастолическую функцию, провоцирование сердечной недостаточности, фиброза миокарда, уменьшение уровня циркулирующего эритропоэтина, влияние на молекулярные сигнальные пути и активацию воспаления [15].
Железодефицитная анемия является самой распространенной формой в популяции, однако у пациентов кардиологического профиля уступает первенство АХЗ и составляет до 21% [16].
Распространенность железодефицитного состояния при ХСН во многом зависит от критериев определения. Если учитывать лишь снижение насыщения трансферина менее 16%, то его можно обнаружить у 78% анемичных и 61% неанемиченых больных ХСН, если к критериям добавить уровень ферритина 30-100 мг/л, то распространенность снизится до 20 и 15% соответственно [17]. В другом исследовании, где критериями железодефицита были значения ферритина менее 100 мг/л при насыщении трансферина менее 16%, нарушения выявлены у 61% анемичных и 43% неанемичных пациентов ХСН [18].
Таким образом, можно говорить о высоком распространении у больных ХСН как абсолютного (определяемого как уровень ферритина 100 мг/л и процента сатурации трансферина 200 мкг/л.
Разграничение АХЗ и ЖДА имеет важное практическое значение: некорректная трактовка пациента с АХЗ как имеющего дефицит железа влечет за собой неэффективную терапию железом с риском развития осложнений (перегрузки железом, особенно при внутривенном введении). Показатели дифференциальной диагностики при АХЗ, ЖДА и их сочетаниях представлены в таблице.
Показатели дифференциальной диагностики при АХЗ и ЖДА
| Показатели | ЖДА | АХЗ |
| Сывороточное железо | Снижено | Снижено |
| ОЖСС | Повышена | Снижена или N |
| Средний корпускулярный объём | Снижен | Снижен (30%) |
| Насыщение трансферрина | Снижено | N или снижено |
| Сывороточный ферритин | Снижен | N |
| Железо костного мозга | Снижено или отсутствует | N |
| Тест с пероральным железом | Подъем hb | Нет подъема Hb |
Анемия при почечной недостаточности
У больных с хроническими заболеваниями почек наиболее важный вклад в развитие анемии вносят снижение продукции эритропоэтина вследствие уменьшения массы функциональных тканей почек и антипролиферативное действие уремических токсинов. Кроме того, развитие анемии может быть вызвано сокращением продолжительности жизни эритроцитов со 120 дней до 70-80 дней, а также потерей крови, ингибированием эритропоэза в результате хронического воспаления, недостатком свободного железа в организме и дефицитом нутриентов, побочным действием лекарств. По современным представлениям, в случае ХПН можно говорить о едином патогенетическом механизме с АХЗ [20].
У большинства пациентов с ХСН и анемией имеется хроническая болезнь почек (ХБП) различной степени, то есть снижение скорости клубочковой фильтрации (СКФ) 2 . Такое сочетание анемии, ХБП и ХСН D.S.Silverberg и соавт. предложили называть кардиоренальным анемическим синдромом, каждое из трех составляющих которого ухудшает течение остальных двух (рис. 3) [21].
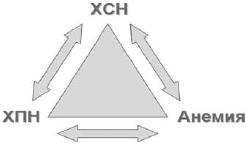
Рис 3. Взаимосвязь при кардиоренальном анемическом синдроме (модифицированна по [21])
Действие лекарств
Применительно к кардиологической практике можно выделить три основных лекарственных воздействия, которые могут провоцировать возникновение и поддержание анемии:
1. Непосредственное подавление костного мозга (вплоть до апластической анемии) могут вызывать следующие лекарственные препараты: НПВС, цитостатики, мерказолил, метамизол (анальгин).
2. Невозможность восстановления присутствующего в пище трехвалентного железа до двухвалентного (всасывающегося во много раз быстрее, нежели трехвалентное) в связи с относительной гипоацидностью (обусловленной сопутствующим применением антисекреторных или антацидных препаратов).
3. Ингибиторы ангиотензинпревращающего фермента и антагонисты к рецепторам ангиотензина могут уменьшать продукцию эритропоэтина и чувствительность к нему костного мозга, так как ангиотензин является мощным стимулятором синтеза эритропоэтина и эритропоэза [22].
Кроме того, необходимо помнить, что нитраты способны вызывать метгемоглобинемию и привести к снижению кислородной емкости крови, однако этот эффект наблюдается в основном при использовании очень высоких доз [23].
Анемия — фактор риска ССЗ
Наличие анемии у пациентов само по себе ассоциировано с пожилым возрастом, нарушением функции почек, сахарным диабетом, тяжелой сердечной недостаточностью, снижением переносимости физических нагрузок и низкими показателями качества жизни [24]. В этой связи наибольшую ценность представляют исследования, выполненные на больших выборках, позволяющие при анализе провести коррекцию на другие факторы риска.
Из исследований, проведенных на популяционном уровне, по данному вопросу можно выделить ARIC-study (Atherosclerosis Risk in Communities), в котором проводилось наблюдение более 6 лет за почти 14 000 пациентов без ССЗ [25]. При оценке анемии как фактора риска ССЗ оказалось, что пациенты со сниженным уровнем гемоглобина имели риск развития указанных заболеваний почти в 1,5 раза выше вне зависимости от всех остальных факторов риска ССЗ (рис. 4).
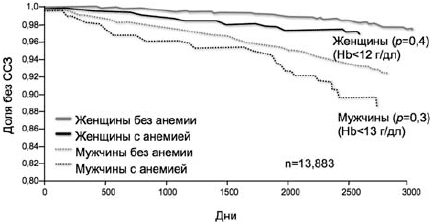
Рис 4. Кривые Каплана-Мейера для ССЗ у мужчин и женщин, стратифицированных по наличию или отсутствию анемии [25]
Анализ других работ по оценке популяционного риска анемии дал противоречивые результаты. Исследователи пришли к выводу о необходимости дополнительных изысканий для определенных рекомендаций в этой области [2].
Анемия и ИБС
Широко известно, что ряд некоронарных заболеваний, в патогенезе которых имеет место гемическая или тканевая гипоксия, может инициировать клинические проявления ИБС, оказывать отягчающее влияние на характер течения основного заболевания. Возможным механизмом представляется усиление симпатической активности и сердечного выброса за счет длительной индуцированной гипоксией вазодилятации, что приводит к гипертрофии левого желудочка и увеличению размеров сердца и, соответственно, к повышению потребления кислорода [26]. Также у пациентов с ИБС по сравнению со здоровыми отмечена сниженная толерантность миокарда к низким уровням гемоглобина [27].
Вопросы сочетания ИБС и анемий представлены в научной литературе немногочисленными источниками. При рассмотрении вопроса влияния анемии на исходы ИБС можно выделить три основных направления исследований:
Наличие анемии перед проведением чрескожного коронарного вмешательства у пациентов с инфарктом миокарда (ИМ) с подъемом сегмента ST достоверно увеличивает риск большого кровотечения как на протяжении 30 сут после ИМ, так и в течение 1 года [28]. Анализ более чем 45 000 случаев чрескожного коронарного вмешательства при ОКС и стабильной ИБС показал, что анемия является независимым фактором риска госпитальной летальности у мужчин и серьезных сердечно-сосудистых осложнений и у мужчин, и у женщин [7]. Повышенный риск неблагоприятных исходов также отмечается у пациентов со сниженным уровнем гемоглобина перед аортокоронарным шунтированием [29].
Сходные результаты показывают исследования, посвященные исходам ОКС в зависимости от наличия анемии, в которых снижение уровня гемоглобина признается значительным фактором риска прогрессирования ИБС, развития ХСН и нарушений ритма, а также смертности [30]. N.C.Meneveau и соавт., помимо признания анемии как независимого фактора риска смерти при ОКС, предлагают включить ее наравне с другими факторами в шкалу риска GRACE (Global Registry of Acute Coronary Events) для более точного прогноза [31].
Исследования, проведенные на популяционном уровне [32] и у больных ИБС [33], подтверждают наличие U-образной зависимости между уровнем гемоглобина и сердечно-сосудистой заболеваемостью и смертностью, т. е. высокий уровень гемоглобина (>13 г/дл) ассоциирован с плохим прогнозом, наряду с низким уровнем.
Анемия и ХСН
Факт увеличения общей и сердечно-сосудистой смертности при наличии анемии у больных с ХСН подтвержден в большом количестве исследований. Так, в ретроспективном исследовании SOLVD показано, что снижение гематокрита на 1% увеличивает общую смертность больных ХСН на 2,7% [34]. Исследование OPTIME продемонстрировало увеличение риска смерти или повторной госпитализации на 12% при уровне гемоглобина менее 12 г/дл [35]. При этом более тяжелый функциональный класс СН по NYHA ассоциировался с более низким уровнем гемоглобина и высоким уровнем креатинина. Имеются данные о наиболее худшем прогнозе относительно сердечно-сосудистой смертности при ЖДА по сравнению другими видами анемии [36].
Анемия при ХСН также является независимым фактором риска более тяжелого течения заболевания: высокого функционального класса, сниженной переносимости физических нагрузок, когнитивных нарушений, низкого качества жизни [37].
Анемия и гипертоническая болезнь
Данные о соотношениях анемии с гипертонической болезнью, несмотря на их широкое распространение, достаточно немногочисленны. Имеются указания на связь анемии с повышением артериального давления в ночные часы и среднего артериального давления (сумма диастолического давления плюс 1/3 пульсового) по данным суточного мониторирования [38], а также на достоверно более широкую распространенность нормоцитарной анемии у пациентов с неконтролируемым уровнем артериального давления — 20%, по сравнению с пациентами поддерживающих нормотонию — 16% (р=0,03) [39].
Лечение
Терапевтические мероприятия, направленные на устранение анемии, должны быть обращены, в первую очередь, на устранение этиологического фактора. В соответствие с этим пациенты должны проходить полноценное обследование для выяснения причины анемии. В рамках данного обзора мы остановимся на АХЗ и ЖДА, составляющих по отдельности или в сочетании подавляющее большинство анемий в кардиологической практике. В данных ситуациях в качестве основной терапии применяются пероральные и внутривенные препараты железа, а также препараты эритропоэтина.
Препараты железа
В случаях выявления устранимой причины ЖДА лечение должно быть направлено на устранение этиологического фактора (эрозивно-язвенные и опухолевые поражения ЖКТ, миома матки, энтериты, алиментарная недостаточность и др.). В этом случае, а также при наличии неустранимой причины непроходимо проводить патогенетическую терапию препаратами железа (ПЖ).
Необходимо отметить ошибочность мнения о возможности коррекции дефицита железа с помощью пищевых продуктов с высоким содержанием железа. Предвидя установление данного факта, врач Мелампас (Melampus) в Греции за 1500 лет до н. э. для избавления принца Ификласа Тезалия (Iphyclus of Thesaly) от полового бессилия, возникшего у него на почве постгеморрагической анемии, давал ему вино с ржавчиной, соскобленной с лезвия старого ножа [40].
В настоящее время мы имеем широкий выбор препаратов железа (ПЖ) для приема внутрь, которые назначаются в большинстве случаев (при отсутствии специальных показаний). Основные ПЖ в виде солей представлены сульфатом, глюконатом, хлоридом, фумаратом, глицинсульфатом. Среди ПЖ в виде железосодержащих комплексов, обладающих большей степенью абсорбции, имеются железо-полимальтозный комплекс, железо-сорбитоловый комплекс, протеин сукцинилат железа, железо-сахаратный комплекс.
Необходимо учитывать, что всасывание железа может уменьшаться под влиянием содержащихся в некоторых пищевых продуктах веществ — фитинов (рис, соя), фосфатов (рыба, морепродукты), танин (чай, кофе). Препараты железосодержащих комплексов (в частности, гидрокси-полимальтозный комплекс) лишены подобных недостатков, поскольку пищевые продукты и медикаменты не оказывают влияния на всасываемость железа в виде трехвалентной формы.
Оптимальная тактика ведения больных ЖДА предполагает насыщающую и поддерживающую терапию ПЖ. Длительность насыщающей терапии зависит от темпов прироста и сроков нормализации показателей гемоглобина, составляя в среднем 3-4 нед, при этом минимальная суточная доза свободного железа должна составлять не менее 100 мг (оптимальная 150-200 мг). Поддерживающая терапия показана в тех ситуациях, когда сохраняется или трудно устранима причина дефицита железа (меноррагии, беременность, патология кишечника).
Оценка результатов лечения:
1. Изменение содержания ретикулоцитов. Считается, что ретикулоцитарный криз появляется на 3-7 сут от начала лечения препаратами железа. Содержание ретикулоцитов может при этом возрастать до 10-20 промилле. Максимальная ретикулоцитарная реакция наступает на 7-10 сут от начала лечения.
2. Прирост гемоглобина начинается с 5 сут при правильном лечении. Если в течение этого периода прироста гемоглобина нет, то это говорит о плохом усвоении препаратов железа. Нормальным считается прирост гемоглобина 1% в сутки или на 0,15 г/сут.
3. Восстановление числа эритроцитов и цветного показателя.
В большинстве случаев для коррекции дефицита железа при отсутствии специальных показаний ПЖ следует назначать внутрь. Показания для внутривенного введения ПЖ у больных ЖДА определяется конкретной клинической ситуацией, в частности, основными являются: состоянием кишечного всасывания и переносимостью пероральных ПЖ.
При переводе на парентеральный прием обязательно надо контролировать уровень сывороточного железа. Без этого показателя вводить внутривенно препараты железа противопоказано (кроме массивной кровопотери). При переводе с перорального на парентеральный прием, пероральное железо должно быть отменено за 2-3 дня.
Несколько другой взгляд относительно пути введения препаратов железа при ХСН представлен в обзоре D.S.Silverberg и соавт. [41]. При сравнении применения пероральных и внутривенных форм ПЖ оказалось, что при отдельном применении, а также в сочетании с эритропоэтином пероральных форм у больных с ХСН не удается достичь того положительного эффекта, получаемого при внутривенном введении, вероятно, за счет блокирования всасывания железа гепсидином. Данное заключение стало возможно после ряда исследований в этой области, которые начались с в некотором смысле революционного подхода в лечении пациентов с ХСН и дефицитом железа, предложенного английскими исследователями, по аналогии с лечением у больных с почечной патологией — внутривенного введения препаратов железа вне зависимости от наличия анемии без эритропоэтина [42]. В данном исследовании, а также в двух других[43, 44] показано достоверное увеличение уровня гемоглобина, фракции выброса левого желудочка, функционального класса ХСН, качества жизни, функции почек, снижение натрийуретических пептидов, С-реактивного белка, а также уменьшение частоты госпитализаций.
В другом исследовании терапии внутривенным железом при ХСН у больных с дефицитом железа вне зависимости от наличия анемии показано улучшение функционального класса ХСН, потребления кислорода и общего состояния даже при отсутствии повышения гемоглобина [45]. Эти данные подтверждают возможность непосредственного влияния железа на митохондриальные процессы окисления. Учитывая существенные различия по клинико-фармакологическим параметрам между препаратами железа необходимо отметить, что в данных исследованиях применялось железо в форме гидроксидсахарозного комплекса (венофер), по применению которого у больных ХСН накоплена в настоящий момент наибольшая доказательная база.
На этом фоне интересным представляется появление в распоряжении врачей нового препарата железа в виде карбоксимальтозатного комплекса (феринъект), обладающего существенно более удобным режимом введения (1 раз в неделю) и лучшим профилем безопасности по отношению к другим препаратам железа. Данная форма железа изучалась в одном из недавних крупных исследований, аналогичном упомянутым выше, у пациентов с ХСН и дефицитом железа вне зависимости от наличия анемии -FAIR-HF, показавшем при 6-месячном наблюдении достоверный переход в более низкий функциональный класс ХСН и улучшение показателей качества жизни при применении внутривенного препарата железа вне зависимости от снижения уровня гемоглобина [46]. Результаты данной работы позволяют частично ответить на вопрос о первичной роли железодефицита по сравнению с наличием анемии в патогенезе ХСН и необходимости его максимально безопасной коррекции.
В настоящий момент отсутствуют полноценные данные о влиянии монотерапии внутривенными препаратами железа на смертность и другие неблагоприятные исходы ХСН при долговременном наблюдении, для окончательного решения этого вопроса необходимы длительные крупные исследования, по результатам которых будут внесены изменения в соответствующие рекомендации.
Эритропоэтины
Применение рекомбинантного эритропоэтина и его в 3 раза более длительно действующего деривата дарбепоэтина в кардиологии наиболее всего изучено у пациентов с ХСН. В нескольких небольших исследованиях применения эритропоэтина в качестве монотерапии или в сочетании с парентеральными препаратами железа было показано снижение смертности и частоты госпитализаций [47]. Наряду с этим показан положительный эффект от данной терапии на различные клинические и функциональные показатели: систолическую и диастолическую функцию правого и левого желудочка, дилатацию камер сердца, гипертрофию левого желудочка, функциональный класс сердечной недостаточности, переносимость физических нагрузок, потребление кислорода, калорийность потребляемой пищи, качество жизни, активность эндотелиальных прогениторных клеток [47]. Данные эффекты не могут полностью объяснены воздействием на эритропоэз и связываются с плеотропными эффектами эритропоэтина, в частности активацией эндотелиальной NO синтетазы и AKT (протеинкиназа В), которая медиирует фосфорилирование, приводя к длительной NO-зависимой вазодилатации [48].
Эритропоэтины, позволяющие увеличить уровень гемоглобина в среднем на 2 г/дл, считаются основными препаратами для коррекции выраженной анемии, в том числе из-за редкости возникновения побочных реакций. Однако данные полученные в онкологических исследованиях свидетельствуют о повышенной частоте сердечно-сосудистых неблагоприятных исходов (в основном за счет тромботических осложнений) при превышении уровня гемоглобина более 12 г/дл [49]. При этом стоит отметить, что дозы эритропоэтинов в данных исследованиях в несколько раз превышали, используемые при ХСН. С другой стороны, у пациентов с ХСН и ХПН не выявлено дополнительного преимущества повышения уровня гемоглобина выше 11-12 г/дл, более того отмечено повышенное количество неблагоприятных исходов при повышении уровня гемоглобина выше 13 г/дл, так называемая U-образная зависимость уровня гемоглобина и смертности [50]. В настоящий момент, в отсутствии официальных рекомендаций по целевому уровню гемоглобина при ХСН, большинство исследователей сходятся на значении 12 г/дл [47].
Заключение
В настоящее время существуют достоверные данные о необходимости активного выявления и коррекции анемии у пациентов кардиологического профиля. Особый интерес вызывает вопрос терапии анемии при длительно протекающей сердечно-сосудистой патологии, в частности ХСН. Накопленная доказательная база у данных больных по лечению анемии эритропоэтинами и/или препаратами внутривенного железа не позволяет однозначно определить целесообразность и безопасность такого подхода. Данные исследований по применению внутривенных препаратов железа в качестве монотерапии у больных ХСН и железодефицитом вне зависимости от наличия анемии могут существенно расширить показания для их применения при данной патологии. Этот и некоторые другие вопросы, как например, наличие универсального маркера ответа при терапии препаратами железа, целевых уровнях гемоглобина при различных заболеваниях и многие другие требуют своего разрешения в научных работах.
До получения результатов крупных исследований лучшим подходом остается считать применение пероральных препаратов железа у пациентов с умеренной ЖДА, а у больных с тяжелой анемией возможна комбинация внутривенного железа и препаратов эритропоэтина, что позволит уменьшить дозы и снизить частоту побочных эффектов. При ХСН даже только с дефицитом железа без анемии можно рассмотреть назначение внутривенных препаратов железа.
источник
Основными факторами, влияющими на обмен железа, являются потребности гемопоэза, пищевой фактор и уровень запаса металла в тканях. Количество железа в организме поддерживается тремя основными путями:
1. Регуляция всасывания в кишечнике.
2. Поддержание рециркуляции железа эритроцитов.
3. Регуляция количества поступающего в клетку железа.
Среди белков-регуляторов обмена железа к настоящему времени наиболее хорошо изучен гепсидин (hepcidin), олигопептид из 25 аминокислот.
В норме гепсидин синтезируется в печени при достаточном количестве железа в гепатоците и особенно при его перегрузке железом . Эффектом пептида в клетках-мишенях является интернализация и деградация белка ферропортина , отвечающего за выход ионов железа в кровь:
- в энтероците действие гепсидина приводит к тому, что большая часть железа остается в клетке, запасается в ферритине и теряется при слущивании кишечного эпителия,
- при действии гепсидина на макрофаги и гепатоциты происходит задержка железа внутри этих клеток.
При патологических состояниях гепсидин повышается в крови на фоне воспалительных процессов и бактериальных инфекций, т.к. он является белком острой фазы. Усиление синтеза происходит под действием провоспалительных цитокинов (IL-6, IL-1α, TNF-α), наиболее эффективным из которых является интерлейкин-6 . Образующийся избыточный гепсидин, уровень которого может повышаться в сотни раз (!), блокирует выход железа из макрофагов, гепатоцитов и энтероцитов, вызывая тем самым гипоферремию. Недостаток железа в крови приводит к снижению эритропоэза – развивается «анемия воспаления» («анемия хронических заболеваний»).
Регуляцию поступления железа в клетку обеспечивает система IRE/IRP (англ. IRE, iron-responsive element — железочувствительный элемент и IRP, iron-responsive element-binding proteins — белок, связывающийся с железочувствительным элементом). Основой регуляции является наличие особого участка на матричной РНК — железочувствительного элемента IRE, связывающего специфичный к нему белок IRP.
В данном случае используется способность молекул IRP связываться с участком IRE соответствующих мРНК для двух белков – рецептора трансферрина и ферритина. При этом в комплексе с железом IRP не активен и не присоединяется к мРНК, без железа — может присоединяться.
1. Присоединение IRP к мРНК рецептора трансферрина ближе к ее 3′-концу защищает мРНК от разрушения РНКазами, действующими с 3′-конца мРНК:
- при низкой концентрации железа в клетке белок IRP является активным, присоединяется к мРНК и, как следствие, мРНК рецепторов трансферрина существует дольше, образуется больше белков-рецепторов и повышается поток железа в клетки.
- при высоком содержании железа в клетке белок IRP присоединяет железо, становится неактивным, с мРНК связаться не может и, соответственно, не защищает ее от разрушения. Синтез рецепторов к трансферрину не происходит, дополнительное железо клеткой не захватывается.
2. Взаимодействие IRP с мРНК ферритина происходит ближе к 5′-концу, т.е. там где начинается трансляция:
- при низком содержании железа в клетке присоединение «пустого» активного белка IRP к мРНК для ферритина со стороны 5′-конца не позволяет ей участвовать в процессе трансляции и синтезе новых молекул ферритина,
- когда концентрация железа в клетке возрастает, оно присоединяется к IRP, снижает его сродство к мРНК и позволяет синтез ферритина.
Конечным результатом при наличии железа в клетке является исчезновение рецепторов к трансферрину с мембраны и увеличение синтеза молекул ферритина, депонирующего железо. При отсутствии железа активируется синтез рецепторов к трансферрину и захват железа из крови, и подавление синтеза запасающего белка ферритина.
источник