 Анемия Под определение «гемолитическая анемия» подпадает любая анемия, характеризующаяся преобладанием гемолиза (процессом распада эритроцитов) над эритропоэзом (процессом образования эритроцитов). Гемолиз может быть иммунный и неиммунный. Гемолитическая анемия бывает наследственные и приобретенные.
Анемия Под определение «гемолитическая анемия» подпадает любая анемия, характеризующаяся преобладанием гемолиза (процессом распада эритроцитов) над эритропоэзом (процессом образования эритроцитов). Гемолиз может быть иммунный и неиммунный. Гемолитическая анемия бывает наследственные и приобретенные.
- Наследственная форма гемолитической анемии, обусловленная нарушением мембраны эритроцитов
- Наследственная форма гемолитической анемии, обусловленная нарушением активности ферментов эритроцитов
- Наследственная форма гемолитической анемии, обусловленная нарушением синтеза или структуры гемоглобина
- Анемия, обусловленная влиянием антител
- Анемия, обусловленная изменением структуры мембраны, вызванной соматической мутацией
- Анемия, обусловленная механическим повреждением оболочки эритроцитов
- Анемия, вызванная химическим повреждением эритроцитов
- Анемия, вызванная дефицитом витаминов (фолиевой кислоты и цианокобаламина)
- Анемия, вызванная разрушением эритроцитов паразитами
Болезнь Минковского-Шоффара (наследственный микросфероцитоз) – группа наследственных гемолитических анемий, характеризующихся образованием микросфероцитов (шаровидных эритроцитов) и обусловленных дефектом протеинов цитоскелета эритроцитов. При этом эритроциты теряют часть мембраны, уменьшается соотношение площади поверхности к объему, в результате чего эритроцит превращается в микросфероцит. Как правило, патология наследуется по аутосомно-доминантному признаку. Распространенность наследственного микросфероцитоза составляет примерно 1 случай на 1000-4500 человек.
При наследственном микросфероцитозе генетические нарушения влияют на протеины цитоскелета, преимущественно на те, которые объединяют цитоскелет с мембраной эритроцита. У большинства больных отмечается значительный дефицит спектрина, и только в некоторых случаях этот дефицит обусловлен генетическими дефектами самого спектрина.
Главные признаки наследственного микросфероцитоза – анемия, желтуха, спленомегалия (увеличенная селезенка). Анемия возникает из-за внутриклеточного распада эритроцитов. Желтуха развивается посредством непрямой гипербилирубинемии, может быть непостоянной и, как правило, слабо выражена у детей раннего возраста. Повышенное содержание билирубина в желчи часто является причиной образования пигментных желчных камней (даже у детей). Увеличение селезенки (спленомегалия) отмечается практически во всех случаях. При системных инфекционных патологиях интенсивность гемолиза может увеличиваться, в результате чего развивается спленомегалия.
Тяжелые формы наследственного микросфероцитоза характеризуются деформацией скелета: изменение расположения зубов, акрокефалия (башенный череп), высокое верхнее небо, микрофтальмия (уменьшение глазного яблока). В некоторых случаях отмечаются укороченные мизинцы. Могут образовываться трофические язвы на ногах.
Наследственный микросфероцитоз сопровождается апластическими кризами, которые провоцируются инфекцией (особенно парвовирусной).
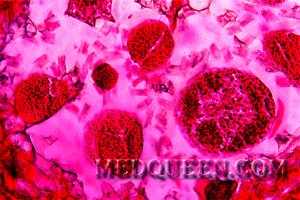
Микросфероцитоз – характерное изменение формы эритроцитов при этой патологии. При анализе мазка крови в биологическом материале наблюдаются микросфероциты в виде мелких клеток без центрального просветления (см рисунок 1). Отметим, что обнаружение микросфероцитов в мазках не всегда является признаком наследственного сфероцитоза.
Рисунок 1. Наследственный микросфероцитоз. Микросфероциты в мазке периферической крови (окр. по Романовскому-Гимзе, ув. ×100)
Такой признак обнаруживается при аутоиммунной гемолитической анемии с неполными тепловыми агглютинами, при наследственных дизэритропоэтической анемии. Средний объем эритроцитов, как правило, остается в норме или незначительно снижен. Показатель среднего содержания гемоглобина в эритроцитах в норме или незначительно повышен. Средняя концентрация гемоглобина в эритроцитах повышена почти у 50% пациентов.
Количественным показателем сферичности эритроцитов является осмотическая устойчивость (она снижена). Уровень ретикулоцитов в крови при гемолитическом кризе может значительно повышаться. Миелограмма показывает резкое раздражение красного ростка. Дифференциальный диагноз проводят с аутоиммунной гемолитической анемией, для которой характерна положительная проба Кумбса, отсутствие этой патологии среди родственников пациента и отсутствие данных о начале заболевания в детском возрасте.
Основной метод лечения анемии при наследственном микросфероцитозе – спленэктомия, с помощью которой устраняется анемия; при этом нельзя устранить морфологический дефект эритроцитов.
Наследственная гемолитическая анемия, обусловленная дефицитом глюкозо-6-фосфат дегидрогеназы эритроцитов – наиболее распространенная ферментопатия эритроцитов из группы ферментопатий пентозофосфатного пути метаболизма глюкозы. Глюкозо-6-фосфатдегидрогеназа эритроцитов – олигомер (в зависимости от условий может быть димер или тетрамер), который состоит из субъединиц с молекулярной массой 56 000 D. По данным ВОЗ (Всемирной организации здравоохранения) во всем мире количество людей, страдающих этой патологией, составляет более 200 млн. Наиболее широкое распространение этого заболевания характерно для Средиземноморского региона (Сицилия, Греция, Сардиния), негроидной расы, жителей Ближнего и Дальнего востока.
Клиническая картина при наследственной форме гемолитической анемии полиморфна: степень тяжести патологии может колебаться от гемолитической анемии, возникающей спонтанно после рождения, до гемолитических кризов. Гемолитический криз, который может провоцироваться метаболическим ацидозом или гипогликемией, развивается за несколько часов. В тяжелых случаях у больного развивается гемоглобинурия и шок. Также наблюдаются желтуха, моча приобретает бурый или черный цвет, одышка, диарея, рвота, снижение артериального давления, развивается тяжелая анемия, увеличиваются печень (гепатомегалия) и селезенка (спленомегалия).
Тяжелый гемолитический криз может спровоцировать развитие ДВС-синдрома (диссеминированного внутрисосудистого свертывания крови). Некоторые пациенты не переносят конские бобы (Viciafaba), после употребления которых происходит молниеносное развитие гемолитического криза (это явление также известно, как фовизм или примахиновая анемия).
Дефицит глюкозо-6-фосфат дегидрогеназы эритроцитов необходимо подозревать во всех случаях острого гемолиза, особенно у лиц негроидной расы и жителей средиземноморского региона. Диагноз подтверждается путем проведения лабораторных анализов. Острый гемолиз характеризуется быстрым снижением гематокрита с одновременным повышением уровня гемоглобина и непрямого гемоглобина, а также снижением уровня гаптоглобина. Анализ мазка крови показывает наличие фрагментов эритроцитов. Основой диагностики считается качественное (при необходимости – количественное) определение активности глюкозо-6-фосфат дегидрогеназы эритроцитов. У пациентов с вариантом «А-» явление аномального гемолиза проходит, как правило, самостоятельно – такие больные не нуждаются в специальном лечении. В случае развития тяжелого гемолитического криза необходимо проводить форсированный диурез, профилактику ДВС-синдрома, плазмаферез (с целью удаления продуктов гемолиза).
В случае возникновения качественной гемоглобинопатии происходит изменение аминокислотной последовательности цепей глобина. Талассемия (количественная гемоглобинопатия) характеризуется снижением образования цепей глобина без изменения их цепей. Нужно отметить, что разница между качественной и количественной гемоглобинопатиями не абсолютна.
Талассемия (анемия Кули) – группа патологий, обусловленных генетическим нарушением синтеза одной из цепей глобина. В норме процесс синтеза глобиновых цепей сбалансирован, поэтому свободных цепей глобина нет. В случае нарушения синтеза одной из цепей глобина баланс нарушается, образуются лишние цепи, которые агрегируют и откладываются в эритрокариоцитах. Среди жителей Средиземноморья наиболее распространена β-талассемия.
«Большая талассемия» (болезнь Кули, β-талассемия) – наследственная гемолитическая анемия, впервые описанная американскими педиатрами-гематологами Томасом Бентоном Кули (Thomas Benton Cooley) и Ли (P. Lee) в статье «Серия случаев спленомегалии у детей с анемией и необычными изменениями костей» («A Series of Cases of Splenomegaly in Children, with Anemia and Peculiar Bone Changes»), где были приведены случаи у выходцев из стран Средиземноморья. Для анемии Кули характерна тяжелая степень течения с самого детства, задержка роста и изменения костей в результате увеличения объема костного мозга, возникающие в случае отсутствия соответствующего лечения). Также при этой патологии у больного наблюдаются гепатомегалия, спленомегалия, гиперспленизм, деформации черепа (монголоидное лицо, башенный череп); желтуха, бледность и отложение меланина придают коже особый медный оттенок. Кроме этого, наблюдается перегрузка железом сердца, легких, печени, поджелудочной железы и других органов эндокринной системы, переломы костей, сдавления периферических нервов, разного рода инфекционные осложнения.
Результаты лабораторных исследований периферической крови показывают гипохромную анемию, ретикулоцитоз, мишеневидные эритроциты (см рис 2-4).
Рисунок 02. Анемия Кули (большая талассемия). Периферическая кровь. Микроцитоз, выраженная гипохромия, мишеневидные нормобласты и эритроциты (окр. по Романовскому-Гимзе, ув. ×100)
Рисунок 03. Анемия Кули (большая талассемия). Периферическая кровь (окр. по Романовскому-Гимзе, ув. ×50)
Рисунок 04. Анемия Кули (большая талассемия). Периферическая кровь. Множественные мишеневидные эритроциты (окр. по Романовскому-Гимзе, ув. ×100)
Миелограмма демонстрирует раздражение «красного ростка» и повышение количества сидеробластов. Также наблюдается повышение осмотической резистентности эритроцитов и количества билирубина за счет непрямой фракции. В крови повышается содержание железа и ферритина, развивается гемосидероз (чрезмерное отложение гемосидерина в тканях) внутренних органов. При гомозиготной β-талассемии необходимо проводить пренатальную диагностику – забор клеток плода из амниотической жидкости на предмет выявления мутации генов, отвечающих за кодирование β-цепи глобина, с применением метода полимеразной цепной реакции.
Без соответствующего лечения больные анемией Кули умирают в детском возрасте. Продлить жизнь, предупредить деформации костей и задержку роста можно путем регулярных трансфузий эритроцитарной массы (лучше переливать отмытые или размороженные эритроциты) при условии поддержания достаточно высокого уровня гемоглобина. В случае значительной спленомегалии и явлениях гиперспленизма больному показана спленэктомия (удаление селезенки). С целью предотвращения развития гемосидероза пациентам периодически назначают Деферазирокс (Эксиджад) или Дефероксамин (Десферал). Излечение возможно при аллогенной трансплантации костного мозга.
Серповидноклеточная анемия обусловлена носительством гемоглобина, который меняет свою структуру в условиях гипоксии. Самой распространенной аномалией структуры гемоглобина является гемоглобинопатия Sα2β26 глу+вал. При гомозиготном носительстве можно говорить о серповидноклеточной анемии; при гетерозиготном носительстве – серповидноклеточная аномалия. Патология наследуется по аутосомно-доминантному признаку. При серповидноклеточной анемии наблюдается мутация, в результате которой в цепи глобина глутаминовая кислота заменяется валином. В результате растворимость гемоглобина S при отдаче кислорода снижается, что приводит к образованию геля.
Серповидноклеточная анемия наиболее распространена среди населения Центральной Африки, Турции, Индии, Кубы. У больных диагностируется анемия, тромботические осложнения, поражения костей и суставов (отмечаются некрозы плечевой и бедренной костей). Кроме этого, тромбозы осложняются инфарктами (сердца, легких, почек, селезенки, головного мозга), приступами сильной боли в области живота. У детей отмечаются нарушения физического (отставание в росте) и полового развития, ночное недержание мочи, нарушение зрения (тромбозы сосудов сетчатки). Также могут развиваться гемолитический, апластический и секвестрационные кризы, при этом в селезенке происходит резкое накопление эритроцитов, что вызывает гиповолемический шок и резкое снижение уровня гемоглобина.
Для анализов крови при апластической анемии характерны низкий уровень гемоглобина, наличие серповидных эритроцитов (рисунок 5), базофильная пунктация эритроцитов, их мишеневидность, повышение уровня ретикулоцитов и непрямого билирубина. Миелограмма демонстрирует раздражение «красного ростка».
Рисунок 5. Серповидноклеточная анемия. Периферическая кровь. Серповидные и мишеневидные эритроциты. выраженная гипохромия эритроцитов (окр. по Романовскому-Гимзе, ув. ×100)
В качестве лечения применяют адекватную инфузионную терапию, переливания эритроцитарной массы, оксигенотерапии.
К приобретенным гемолитическим анемиям относится группа заболеваний разного патогенеза, которые объединяет внутрисосудистый гемолиз (гемолиз эритроцитов в периферической крови). В зависимости от механизма эритролиза приобретенная гемолитическая анемия может носить иммунный и неиммунный характер. Но, несмотря на разные патогенетические механизмы, клинические признаки этих анемий часто совпадают.
Гемолитическая анемия у пациентов с протезированными клапанами сердца и сосудами развивается примерно в 10% случаев при протезированном аортальном клапане. При использовании стеллитовых запирательных элементов частота гемолиза незначительно увеличивается (по сравнению с селиконовыми). Также некоторое увеличение частоты гемолиза отмечается при наличии околоклапанной регургитации и при малом диаметре клапана. Биопротезы (свиные клапаны) в редких случаях являются причиной механического гемолиза. Гораздо реже причиной гемолиза может быть также протезированный митральный клапан, так как трансклапанный градиент давления в этом случае ниже.
Гемолиз протезированными клапанами происходит в результате одновременного действия сразу нескольких факторов:
- Значительная сила сдвига, которая при турбулентном токе крови действует на мембрану эритроцитов, особенно когда под высоким давлением кровь проходит через маленькое отверстие (например, при околоклапанной регургитации)
- Отложения фибрина на участках неплотного прилегания кольца клапана к тканям сердца
- Прямое механическое повреждение эритроцитов при закрытии запирательного элемента
Значительное разрушение эритроцитов может наблюдаться после закрытия дефекта межпредсердной перегородки типа ostium primum заплатой из синтетического материала. Умеренное сокращение жизни эритроцитов с легкой анемией или без нее может наблюдаться при значительном обызвествлении аортального клапана. Механический гемолиз обнаруживается также у пациентов, перенесших аортокоронарное и аортобедренное шунтирование.
Тяжелые случаи механического гемолиза сопровождаются тяжелой анемией, ретикулоцитозом, обнаруживаются фрагментированные эритроциты (шизоциты), гемоглобинемия и гемоглобинурия, повышается активность лактатдегидрогеназы, снижается уровень гаптоглобина. Выведение железа из организма с мочой в виде гемосидерина или гемоглобина может вызвать дефицит железа в организме. В случае развития дефицита железа пациенту назначается пероральный прием препаратов железа. Терапия препаратами железа способствует повышению уровня гемоглобина и способствует снижению сердечного выброса и снижению интенсивности гемолиза. Отметим, что ограничение физической активности также способствуют снижению интенсивности распада эритроцитов. Если предпринимаемые меры не приводят к желаемому результату, нужно полностью устранить околоклапанную регургитацию или заменить протез.
источник
Наследственная сфероцитарная гемолитическая анемия (болезнь Минковского — Шоффара; врожденная микросфероцитарная гемолитическая анемия, врожденный сфероцитоз)
Наследственная сфероцитарная гемолитическая анемия впервые наиболее обстоятельно описана в 1900 г. Minkowsky и в 1907 г. Chauffard. В основе заболевания лежит генетический дефект эритроцитов, приводящий к изменению их формы — к сфероцитозу и к снижению минимальной осмотической резистентности к гипотоническим растворам поваренной соли.
Патогенез болезни до сих пор остается недостаточно ясным. До конца неизвестна истинная причина и характер внутриклеточного дефекта эритроцитов (Aussannaire с соавт., 1960). Предположение о врожденном нарушении метаболизма эритроцитов в результате ферментных дефектов высказывают Dacie (1954), Prankerd с соавт. (1955, 1960). Имеются работы, указывающие на недостаток энзима энолазы в эритроцитах больных (Motulsky с соавт., 1958). Нарушение нуклеотидного состава красных кровяных телец отмечали Corsini с соавт. (1967), Mircevova с соавт. (1967).
Изменение обмена дезоксинуклеазы у детей со сфероцитарной гемолитической анемией наблюдал Willy (1967). Исследования Shojania с соавт. (1964) свидетельствуют о снижении уровня фолиевой кислоты у больных с данным заболеванием. Работы Jacob (1967, 1968) указывают на увеличение проницаемости оболочки сфероцитов для ионов натрия. Повышенная проницаемость натрия через оболочку эритроцитов сопровождается потерей их осмотической устойчивости в результате снижения концентрации ионов калия и выхода липидов.
Имеется предположение, что сфероцитоз эритроцитов при данном заболевании является результатом нарушения в них транспорта катионов, связанный с изменением контрактильных свойств мутантного белка, содержащегося в мембране эритроцитов и обладающего АТФазной активностью.
Применяя метод кислотных эритрограмм, М. Д. Бриллиант и А. И. Воробьев (1962) выявили нарушение проницаемости мембраны эритроцитов и изменение их липидного состава у взрослых больных со сфероцитарной гемолитической анемией. Подобное наблюдали и Cooper, Sande (19ЬУ). Сферуляцию эритроцитов А. И. Осипов (1965) связывает с «поломом» в системе АТФ — АТФазы, поскольку АТФ способствует сохранению формы эритроцитов и поддерживает в них баланс Na и К (Schrier, 1967). Лабораторные исследования Mochler (1967) определяют in vitro снижение гемолиза сфероцитов при добавлении к ним АТФ, глюкозы, глютатиона.
На повышение АТФазной активности при сфероцитарной гемолитической анемии указывают В. Г. Соловьев, Н. П. Ша балов (1970). Данный факт они связывают с наследственным дефектом обмена липидов в мембране эритроцитов. Повышение активности глюкозо6фосфатдегидрогеназы и транскеталазы наблюдал Ю. Р. Ковалев (1970). Наибольшую активность глюкозо6фосфатдегидрогеназы Б. Я. Резник и Ю. А. Сорока отмечают во время криза. Высокую активность гексокиназы у больных со сфероцитарной гемолитической анемией наблюдал Ю. А. Сорока (1973).
Внутриклеточные молекулярные изменения, происходящие при наследственной сфероцитарной гемолитической анемии, способствуют сферуляции эритроцитов, что сопровождается уменьшением их диаметра, увеличением толщины и объема. Цикл жизни сфероцитов укорачивается. Длительность их жизни, определяемая методом Эшби (1919) и с помощью радиоактивных изотопов, сокращается иногда до 18 дней (Я. Д. Сахибов, 1967) при норме 90 — 120 дней.
Наблюдения Э. Г. Метровелли (1966) и Ф. Б. Репиной (1968) выявили укорочение длительности жизни эритроцитов иногда до 1 — 1,5 дня у детей, больных наследственной сфероцитарной гемолитической анемией.
Длительное время дискутировался вопрос о роли селезенки при данном заболевании. Minkowsky (1900), Eppinger, Charnas с соавт. (1913), Heilmeyer (1950) и др. основное значение придавали селезенке в развитии гемолитического процесса, считая, что селезенка способствует сферуляции и разрушению эритроцитов. Свое предположение они основывали на том, что удаление селезенки приводит к выздоровлению больных. Однако последующие наблюдения опровергли эту точку зрения. Перекрестное переливание эритроцитов от больных сфероцитарной гемолитической анемией здоровым указали на сокращение цикла жизни сфероцитов в их крови и нормальную длительность жизни эритроцитов донора в крови больных сфероцитарной гемолитической анемией. Эти опыты указывают на непричастность в данных ситуациях селезенки (Mollison с соавт., 1949; Dacie, 1954).
Исследования Г. А. Алексеева (1970) осмотической резистентности эритроцитов крови селезеночной вены (во время спленэкто мии) показали их более высокую осмотическую резистентность в сравнении с эритроцитами периферической крови, что указывает на отсеивающую роль селезеночного фильтра. В селезенке происходит гемолиз подготовленных к распаду эритроцитов, чему способствует медленное кровообращение в ней, и узкие устья венозных синусов, где происходит трав матизация неполноценных эритроцитов. Все сказанное свидетельствует о том, что основная причина гемолиза эритроцитов при наследственной сфероцитарной гемолитической анемии заключается в самих эритроцитах, в их генетической неполноценности. Роль селезенки в гемолитическом процессе является вторичной. Однако в последние годы появились высказывания о том, что селезенка не является простым «кладбищем» эритроцитов при этом заболевании. Ее роль в патологическом процессе сложнее (Д. Ф. Окунев, 1926; Ю. И. Лорие, 1964).
Остается нерешенным вопрос о месте сферуляции эритроцитов в костном мозге или в периферической крови. Измерение диаметра ретикулоцитов и клеток костного мозга показало их нормальные размеры, что свидетельствует о процессе сферуляции эритроцитов уже по выходе из костного мозга.
При наличии интенсивного гемолиза эритроцитов селезенка гиперплазирует и вторично усиливает проявление основного заболевания, т. е. ведет к возникновению явлений вторичного гиперспленизма. На усиление функции ретикулогистиоцитарной системы при гемолитической анемии указывает Я. Г. Ужанский (1968). Секвестрацию эритроцитов в селезенке при гемолитической анемии наблюдал с помощью радиоизотопов Veeger с соавт. (1961).
Повышенное разрушение эритроцитов в клетках ретикулогистиоцитарной системы селезенки приводит у больных сфероцитарной гемолитической анемией к повышенному освобождению свободного билирубина и накоплению его в крови. Этот пигмент, прокрашивая кожу, слизистые оболочки и внутренние органы, ведет к появлению желтухи. Одновременно со свободным билирубином освобождается железо, в результате чего его количество увеличивается в сыворотке крови, что способствует развитию вторичного гемосидероза органов (В. Я. Орлова, 1962).
Целесообразная реакция организма, стремящегося к компенсации возникших в нем нарушений, выражается у больных в активации деятельности кроветворной системы. Гиперлазирует костный мозг, в крови появляются молодые недостаточно зрелые элементы эритропоэза — ретикулоциты, нормобласты.
«Гемолитические анемии у детей»,
М.Я.Студеникин, А.И.Евдокимова
Сущность лекарственного гемолиза эритроцитов при дефиците Г6ФД заключается в следующем. При встрече эритроцитов с гемолизирующим веществом окислительного действия в норме происходит компенсаторное усиление активности Г6ФД для поддержания редукции глютатиона на необходимом уровне. При наследственном дефиците Г6ФД резервные возможности эритроцитов снижаются и поэтому при встрече их с гемолизирующими агентами резко нарушается процесс восстановления глютатиона, что приводит…
В настоящее время привлекают внимание заболевания, связанные с наследственной недостаточностью ферментов — энзимопатии. Открыто много внутри-эритроцитарных ферментов, недостаток которых может способствовать появлению гемолитической анемии. Ферментопатии можно разделить на две группы. К первой группе относятся дефициты пируваткиназы, глюкозофосфатизомеразы, триозофосфатизомеразы, гексокиназы, фосфофруктокиназы, АТФазы и др. При дефиците ферментов этой группы у больных нарушается течение внутриклеточных гликолитических процессов…
Артур Г., 10 лет. Поступил в клинику 4.1 1971 г. Отец армянин, мать — азербайджанка. У родителей умеренная гипохромная анемия. Мальчик родился весом 3550 г. С раннего возраста страдает экссудативным диатезом. В возрасте 4 лет впервые выявлена анемия. При поступлении в клинику состояние средней тяжести, бледный, склеры субиктеричные, изменения в скелете — «башенный» череп, прогнатизм,…
При гемоглобинозах Н и Zurich чаще всего после дачи лекарственных препаратов развивается гемолитический криз и иногда очень тяжелый. При этом моча может приобретать темный цвет за счет выделения дипирроллов мезабилифусциновой группы. По органам удается выявить спленомегалию. Диагностика гемоглобинопатий группы нестабильных гемоглобинов проводится с помощью лабораторных методов исследования. Они включают достаточно большой арсенал методик, способствующих не…
У большинства гемоглобинов этой группы в каком-то положении р-цепей имеется замена одних аминокислотных остатков на другие. Это приводит к изменению электрофоретической подвижности (преимущественно они мигрируют между гемоглобинами A и А2), нарушению соединения гема с глобином (НЬН Hammersmith, Koln), увеличению образования метгемоглобина (in vivo и in vitro), легкой денатурации белка под влиянием некоторых химических веществ (сульфопрепаратов,…
источник
ЭУМК Педиатрия / 5. Методические пособия / 4 курс Леч / Гемолитические анемии — врожденные и приобретенные
Тема: Гемолитические анемии – врожденные и приобретенные.
Цель изучения: познакомить студентов с понятием гемолитических анемий, рассмотреть различные клинические варианты гемолитических анемий, диагностику, дифференциальную диагностику, осложнения. Изучить изменения в картине крови при различных клинических вариантах гемолитических анемий.
— Понятие гемолитических анемий;
— Классификация наследственных гемолитических анемий;
— Болезнь Миньковского – Шоффара;
— Анемия, связанная с дефицитом Г-6-ФД эритроцитов;
— Классификация приобретенных гемолитических анемий;
— Общие принципы диагностики и лечения гемолитических анемий.
Изложение учебного материала:
Анемия, при которой процесс разрушения эритроцитов преобладает над процессом регенерации, называется гемолитической.
Естественная гибель эритроцита (эритродиерез) происходит спустя 90-120 дней после его рождения в сосудистых пространствах ретикулогистиоцитарной системы, главным образом в синусоидах селезенки и значительно реже непосредственно в кровеносном русле. При гемолитической анемии наблюдается преждевременное разрушение (гемолиз) эритроцитов. Устойчивость эритроцита к различным воздействиям внутренней среды обусловлена как структурными белками клеточной мембраны (спектрин, анкирин, белок 4,1 и др.), так и ее ферментным составом, кроме того, нормальным гемоглобином и физиологическими свойствами крови и других сред, в которых циркулирует эритроцит. При нарушении свойств эритроцита или изменении среды его пребывания, он преждевременно разрушается в кровеносном русле либо в ретикулогистиоцитарной системе различных органов, прежде всего селезенки.
Классификация гемолитических анемий
Обычно выделяют наследственные и приобретенные гемолитические анемии, поскольку они имеют различные механизмы развития и отличаются подходом к лечению. Реже классифицируют гемолитические анемии по наличию или отсутствию иммунопатологии, различая аутоиммунные и неиммунные гемолитические анемии, к которым относятся врожденные гемолитические анемии, приобретенные гемолитические анемии у больных циррозом печени, а также при наличии протезов сердечных клапанов и так называемая маршевая гемоглобинурия.
Гемолитическим анемиям присущ ряд признаков, которые выделяют их из анемий другого происхождения. Прежде всего, это гиперрегенераторные анемии, протекающие с гемолитической желтухой и спленомегалией. Высокий ретикулоцитоз при гемолитическим анемиям обусловлен тем, что при распаде эритроцитов образуются все необходимые элементы для построения нового эритроцита и, как правило, отсутствует дефицит эритропоэтина, витамина В12, фолиевой кислоты и железа. Разрушение эритроцитов сопровождается увеличением содержания в крови свободного билирубина; когда его уровень превышает 25 мкмоль/л, появляется истеричность склер и кожных покровов. Увеличение селезенки (спленомегалия) — результат гиперплазии ее ретикулогистиоцитарной ткани, обусловленной повышенным гемолизом эритроцитов. Общепринятой классификации гемолитических анемий нет.
Наследственные гемолитические анемии.
А. Мембранопатии вследствие нарушения структуры мембраны эритроцита:
Нарушение белков мембраны эритроцитов: микросфероцитоз; эллиптоцитоз; стоматоцитоз; пиропойкилоцитоз.
Нарушение липидов мембраны эритроцитов: акантоцитоз, дефицит активности лецитин-холестерин-ацилтрансферазы (ЛХАТ), увеличение содержания лецитина в мембране эритроцитов, детский инфантильный пикноцитоз.
Дефицит ферментов пентозофосфатного цикла.
Дефицит активности ферментов гликолиза.
Дефицит активности ферментов обмена глутатиона.
Дефицит активности ферментов, участвующих в использовании АТФ.
Дефицит активности рибофосфатпирофосфаткиназы.
Нарушение активности ферментов, участвующих в синтезе порфиринов.
Обусловленные аномалией первичной структуры гемоглобина
Вызванные снижением синтеза полипептидных цепей, входящих в состав нормального гемоглобина
Обусловленные двойным гетерозиготным состоянием
Аномалии гемоглобина, не сопровождающиеся развитием заболевания
Приобретенные гемолитические анемии
А. Иммунные гемолитические анемии:
Гемолитические анемии, связанные с воздействием антител: изоиммунные, гетероиммунные, трансиммунные.
Аутоиммунные гемолитические анемии: с неполными тепловыми агглютининами, с тепловыми гемолизинами, с полными холодовыми агглютининами, связанные с двухфазными холодовыми гемолизинами.
Аутоиммунные гемолитические анемии с антителами против антигена нормоцитов костного мозга.
Б. Гемолитические анемии, связанные с изменением мембран, обусловленные соматической мутацией: ПНГ.
В. Гемолитические анемии, связанные с механическим повреждением оболочки эритроцитов.
Г. Гемолитические анемии, связанные с химическим повреждением эритроцитов (свинец, кислоты, яды, алкоголь).
Д. Гемолитические анемии на фоне дефицита витаминов Е и А.
Е. Гемолитические анемии, обусловленные разрушением эритроцитов паразитами (малярия).
Гемолитические анемии — группа заболеваний, различающихся по своей природе, клинике и принципам лечения, но объединенных единственным признаком — гемолизом эритроцитов. Среди болезней крови гемолитические анемии составляют 5 %, а среди всех анемий гемолитические анемии составляют 11 %. Главным признаком гемолитических состояний является гемолиз — уменьшение продолжительности жизни эритроцитов и их усиленный распад.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ. Физиологическая норма продолжительности жизни эритроцитов колеблется от 100 до 120 дней. Эритроцит имеет мощный метаболизм и несет колоссальную функциональную нагрузку. Обеспечение функций эритроцитов определяется сохранностью структуры и формы клеток и процессов, обеспечивающих метаболизм гемоглобина. Функциональная активность обеспечивается процессом гликолиза, в результате которого синтезируется АТФ, снабжающая энергией эритроцит. Сохранность структуры и нормальный метаболизм гемоглобина обеспечивает структурный белок трипептид-глутатион. Форму поддерживают липопротеиды мембраны эритроцитов. Важным свойством эритроцитов является их способность деформироваться, что обеспечивает свободное прохождение эритроцитов при входе в микрокапилляры и при выходе из синусов селезенки. Деформируемость эритроцитов зависит от внутренних и внешних факторов. Внутренние факторы: вязкость (обеспечивается нормальной концентрацией гемоглобина в средней части эритроцита) и онкотическое давление внутри эритроцита (зависит от онкотического давления плазмы крови, наличия в эритроците катионов магния и калия). При большом онкотическом давлении плазмы ее элементы устремляются внутрь эритроцита, он деформируется и лопается. Нормальное содержание магния и калия зависит от работы транспортного механизма мембраны, который, в свою очередь, зависит от правильного соотношения в мембране белковых компонентов и фосфолипидов, т. е. если нарушается какая-либо часть генетической программы эритроцита (синтез транспортных или мембранных белков), то нарушается равновесие внутренних факторов, что приводит к гибели эритроцита.
Внешние факторы: иммунные факторы, витамины, механизмы, обеспечивающие метаболизм гемоглобина (преимущественно печеночные факторы), транспортные белки, которые обеспечивают перенос гемоглобина, онкотическое давление плазмы крови, паразитарные факторы (малярия). Эритроциты в процессе эволюции стареют и секвестрируются в костном мозге и селезенке. В результате физиологического распада эритроцитов билирубин находится в крови в виде неконъюгированной (свободной) фракции, который доставляется к гепатоцитами, где в результате ферментативной реакции соединяется с глюкуроновой кислотой. Конъюгированная фракция билирубин-глюкуронид из гепатоцитов поступает в желчевыводящие пути и выделяется с желчью в желудочно-кишечный тракт.
При развитии гемолитической анемии продолжительность жизни эритроцитов сокращается до 12—14 дней. Патологический гемолиз делится на внутрисосудистый и внутриклеточный. Внутрисосудистый гемолиз характеризуется повышенным поступлением гемоглобина в плазму и выделением с мочой в виде гемосидерина или в неизмененном виде. Для внутриклеточного гемолиза характерен распад эритроцитов в ретикулоцитарной системе селезенки, что сопровождается повышением содержания свободной фракции билирубина в сыворотке крови, выведением уробилина с калом и мочой, склонностью к холелитиазу и холедохолитиазу.
Болезнь Миньковского—Шоффара (наследственный микросфероцитоз).
Болезнь Миньковского — Шоффара – наследственное заболевание, наследуется по аутосомно-доминантному типу.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ. На практике не наследуется каждый четвертый случай. Очевидно, в основе этого типа лежит некая спонтанно возникающая мутация, сформировавшаяся в результате действия тератогенных факторов. Генетически наследуемый дефект белка эритроцитарной мембраны приводит к избытку в эритроцитах ионов натрия и молекул воды, в результате образуются патологические формы эритроцитов, имеющие сферическую форму (сфероциты). В отличие от нормальных двояковогнутых эритроцитов они не способны деформироваться при прохождении в узких сосудах синусов селезенки. В результате продвижение в синусах селезенки замедляется, часть эритроцитов отщепляется, и образуются мелкие клетки — микросфероциты, которые быстро разрушаются. Обломки эритроцитов захватываются макрофагами селезенки, что приводит к развитию спленомегалии. Повышенное выделение билирубина с желчью обусловливает развитие плейохромии и желчнокаменной болезни. В результате повышенного распада эритроцитов повышается количество свободной фракции билирубина в сыворотке крови, которая выводится из кишечника с калом в виде стеркобилина и частично с мочой. При болезни Миньковского — Шоффара количество выделяемого стеркобилина превышает нормальные показатели в 15—20 раз.
ПАТОЛОГО-АНАТОМИЧЕСКАЯ КАРТИНА. За счет эритроидного ростка костный мозг в трубчатых и плоских костях гиперплазирован, отмечается эритрофагоцитоз. В селезенке наблюдаются уменьшение количества и размеров фолликулов, гиперплазия эндотелия синусов, выраженное кровенаполнение пульпы. В лимфатических узлах, костном мозге и печени может быть выявлен гемосидероз.
КЛИНИКА. В течение заболевания чередуются периоды ремиссий и обострений (гемолитический криз). К развитию гемолитического криза предрасполагают обострение хронической инфекции, интеркуррентные инфекции, вакцинация, психическая травма, перегревание и переохлаждение. В раннем возрасте болезнь обычно выявляется, если подобное заболевание присутствует у родственников. Первый симптом, который должен насторожить, — это затянувшаяся во времени желтуха. Чаще всего первые проявления болезни выявляются у подростков или взрослых людей, так как появляется больше провоцирующих факторов. Вне периода обострения жалобы могут отсутствовать. Период обострения характеризуется ухудшением самочувствия, наличием головокружения, слабости, утомляемости, сердцебиения, повышением температуры тела. Желтуха (лимонно-желтого цвета) является основным и может быть единственным признаком заболевания долгое время. Интенсивность желтухи зависит от возможностей печени конъюгировать свободный билирубин с глюкуроновой кислотой и от интенсивности гемолиза. В отличие от механической и паренхиматозной желтуха гемолитического генеза не характеризуется появлением обесцвеченного кала и мочи цвета пива. В анализе мочи билирубин не выявляется, так как свободный билирубин через почки не проходит. Кал становится темно-коричневым за счет повышенного уровня стеркобилина. Возможна манифестация желчнокаменной болезни на фоне склонности к камнеобразованию с развитием острого холецистита. При закупорке общего желчного протока конкрементом (холедохолитиаз) к клинической картине присоединяются признаки обтурационной желтухи (кожный зуд, билирубинемия, наличие желчных пигментов в моче и т. д.). Характерным признаком наследственного микросфероцитоза является спленомегалия. Селезенка пальпаторно определяется на 2—3 см ниже реберной дуги. При длительном гемолизе спленомегалия выраженная, что проявляется тяжестью в левом подреберье. Печень при отсутствии осложнений обычно нормальных размеров, редко у отдельных больных при длительном течении заболевания она может увеличиваться. Кроме желтухи и спленомегалии, можно отметить расширение границ относительной сердечной тупости, систолический шум, приглушенность тонов. При осмотре могут наблюдаться костные патологии (нарушение роста и расположения зубов, высокое стояние неба, седловидный нос, башенный череп с узкими глазницами) и признаки замедления развития. Уровень гемоглобина обычно не изменен или умеренно снижен. Резкое нарастание анемии наблюдается во время гемолитических кризов. У лиц старшего возраста могут наблюдаться трудно заживающие трофические язвы голени, обусловленные распадом и агглютинацией эритроцитов в периферических капиллярах конечности. Гемолитические кризы появляются на фоне постоянно текущего гемолиза и характеризуются резким усилением клинических проявлений. При этом в связи с массовым распадом эритроцитов повышается температура тела, появляются диспепсические расстройства, боли в животе, нарастает интенсивность желтухи. Провоцируют развитие гемолитических кризов беременность, переохлаждение, интеркуррентные инфекции. В некоторых случаях гемолитические кризы в течение болезни не развиваются.
ГЕМАТОЛОГИЧЕСКАЯ КАРТИНА. В мазке крови микроцитоз, большое количество микросфероцитов. Увеличено также количество ретикулоцитов. Количество лейкоцитов и тромбоцитов в пределах нормы. Во время гемолитических кризов наблюдается нейтрофильный лейкоцитоз со сдвигом влево. В костном мозге наблюдается гиперплазия эритроидного ростка. Билирубинемия не выражена. Уровень непрямого билирубина в среднем составляет 50—70 мкмоль/л. Повышается содержание стеркобилина в кале и уробилина в моче.
ДИАГНОЗ. Диагноз наследственного микросфероцитоза ставят на основании клинической картины, лабораторных исследований. Обязательным является обследование родственников на наличие признаков гемолиза и микросфероцитоза без клинических проявлений.
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА. В периоде новорожденности болезнь Минковского — Шоффара надо дифференцировать с внутриутробной инфекцией, атрезией желчных ходов, врожденным гепатитом, гемолитической болезнью новорожденных. В грудном возрасте — с гемосидерозом, лейкозом, вирусным гепатитом. Острый эритромиелоз нередко путают с гемолитическим кризом, сопровождающимся анемией, лейкоцитозом со сдвигом влево, спленомегалией, гиперплазией эритроидного ростка в костном мозге. Дифференциальная диагностика наследственного микросфероцитоза с аутоиммунными гемолитическими анемиями включает выполнение пробы Кумбса, позволяющей определить антитела, фиксированные на эритроцитах, характерные для аутоиммунных анемий. От наследственного микросфероцитоза необходимо отличать группу несфероцитарных гемолитических анемий. Эти заболевания характеризуются ферментативной недостаточностью в эритроцитах, отсутствием сфероцитоза, нормальной или слегка увеличенной осмотической резистентностью эритроцитов, повышенным аутогемолизом, гипергликемией, не поддающейся коррекции. Часто для дифференциальной диагностики используют кривую Прайса — Джонса (кривую, отражающую размеры эритроцитов), по которой при наследственном микросфероцитозе идет сдвиг к микросфероцитам.
ЛЕЧЕНИЕ. Спленэктомия является единственным эффективным в 100 % случаев методом лечения пациентов с наследственным микросфероцитозом. Несмотря на то, что снижение осмотической резистентности и микросфероцитоз у эритроцитов сохраняются, явления гемолиза купируются, так как в результате спленэктомии удаляется основной плацдарм для разрушения микросфероцитов, при этом исчезают все проявления болезни. Показаниями к спленэктомии являются частые гемолитические кризы, резкая анемизация больных, инфаркт селезенки. Нередко при наличии у больного желчнокаменной болезни симультантно выполняют холецистэктомию. У взрослых больных при легком течении заболевания и компенсации процесса показания к спленэктомии являются относительными. Предоперационная подготовка включает переливание эритроцитарной массы, особенно при выраженной анемии, витаминотерапию. Применение глюкокортикоидных препаратов при лечении наследственного микросфероцитоза не является эффективным.
ПРОГНОЗ. Течение наследственного микросфероцитоза редко бывает тяжелым, прогноз относительно благоприятен. Многие больные доживают до преклонного возраста. Супруги, один из которых болен наследственным микросфероцитозом, должны знать, что вероятность возникновения микросфероцитоза у их детей немногим ниже 50 %.
Наследственные гемолитические анемии, связанные с дефицитом ферментов (ферментопатии).
Группа наследственных несфероцитарных гемолитических анемий наследуется по рецессивному типу. Для них характерны нормальная форма эритроцитов, нормальная или повышенная осмотическая резистентность эритроцитов, отсутствие эффекта от спленэктомии. Дефицит ферментативной активности приводит к повышению чувствительности эритроцитов к воздействию лекарственных средств и веществ растительного происхождения.
Острая гемолитическая анемия, связанная с дефицитом глюкозо-6-фосфатдегидрогеназы (Г-6-ФДГ).
Встречается наиболее часто, по данным ВОЗ, около 100 млн. человек в мире имеют дефицит глюкозо-6-фосфатдегидроге-назы. Дефицит Г-6-ФДГ влияет на синтез АТФ, метаболизм глутатиона, состояние тиоловой защиты. Наиболее широко распространен у жителей средиземноморских стран Европы (Италия, Греция), Африке и Латинской Америке.
ЭТИОЛОГИЯ. Это наследственное заболевание, сцепленное с Х-хромосомой. Некоторые авторы связывают распространение заболевания с малярией. Дефицит Г-6-ФД у больных тропической малярией давал им некоторые преимущества в борьбе с анемией, и она реже погибали. Было обнаружено, что в нормальных эритроцитах паразитов больше, чем в эритроцитах с недостатком Г-6-ФД.
ПАТОГЕНЕЗ. В эритроцитах со сниженной активностью Г-6-ФД уменьшается образование НАДФ и связывание кислорода, а также снижается скорость восстановления метгемоглобина и понижается устойчивость к воздействию различных потенциальных окислителей. Окислители, в том числе и лекарственные, в таком эритроците снижают восстановленный глутатион, что в свою очередь создает условия окислительной денатурации ферментов, гемоглобина, составных компонентов мембраны эритроцитов и влечет внутрисосудистый гемолиз или фагоцитоз. Известно более 40 медикаментов, не считая вакцин и вирусов, которые потенциально способны вызвать острый внутрисосудистый гемолиз у лиц с недостаточной активностью Г-6-ФД. Гемолиз таких эритроцитов могут вызывать также эндогенные интоксикации и ряд растительных продуктов.
Примеры лекарств и продуктов, которые потенциально могут вызывать гемолиз: хинин, делагил, стрептоцид, бактрим, промизол, фурацилин, фуразолидон, фурагин, изониазид, левомицетин, аспирин, аскорбиновая кислота, колхицин, леводопа, невиграмон, метиленовый синий, растительные продукты (конские бобы, горошек полевой, папоротник мужской, голубика, черника).
ПАТОЛОГО-АНАТОМИЧЕСКАЯ КАРТИНА. Наблюдаются иктеричность кожи и внутренних органов, сплено- и гепатомегалия, умеренное набухание и увеличение почек. При микроскопии в канальцах почек обнаруживаются гемоглобинсодержащие цилиндры. В селезенке и печени выявляется макрофагальная реакция с наличием гемосидерина в макрофагах.
КЛИНИКА. Недостаточность Г-6-ФД отмечается преимущественно у лиц мужского пола, имеющих единственную Х-хромосому. У девочек клинические проявления наблюдаются главным образом в случаях гомозиготности.
Выделяют 5 клинических форм недостаточности Г-6-ФД в эритроцитах:
острый внутрисосудистый гемолиз – классическая форма недостаточности Г-6-ФД. Встречается повсеместно. Развивается в результате приема лекарств, вакцинации, диабетического ацидоза, в связи с вирусной инфекцией;
фавизм, связанный с употреблением в пищу или вдыханием цветочной пыльцы некоторых бобовых;
гемолитическая болезнь новорожденных, не связанная с гемоглобинопатией, с групповой и резус-несовместимостью;
наследственная хроническая гемолитическая анемия (несфероцитарная);
Гемолитический криз могут спровоцировать анальгетики, некоторые антибиотики, сульфаниламиды, противомалярийные препараты, нестероидные противовоспалительные средства, химиопрепараты (ПАСК, фурадонин), растительные продукты (стручковые, бобовые) и витамин К, а также переохлаждение и инфекции. Проявления гемолиза зависят от дозы гемолитических агентов и степени дефицита Г-6-ФДГ. Через 2—3 дня после приема препаратов начинаются повышение температуры тела, рвота, слабость, боли в спине и животе, сердцебиение, одышка, нередко развивается коллапс. Моча становится темного цвета (вплоть до черного), что обусловлено внутрисосудистым гемолизом и наличием в моче гемосидерина. Характерный признак внутрисосудистого гемолиза – гипергемоглобинемия, сыворотка крови при стоянии приобретает коричневый цвет за счет образующегося метгемоглобина. Одновременно отмечается и гипербилирубинемия. Повышается содержание желчных пигментов в дуоденальном содержимом, в испражнениях. В тяжелых случаях почечные канальцы закупориваются продуктами распада гемоглобина, снижается клубочковая фильтрация и развивается острая почечная недостаточность. При физикальном исследовании отмечаются иктеричность кожного покрова и слизистых оболочек, спленомегалия, реже увеличение печени. Через 6—7 дней гемолиз заканчивается независимо от того, продолжается ли прием препаратов или нет.
ГЕМАТОЛОГИЧЕСКАЯ КАРТИНА. В течение первых 2—3 дней гемолитического криза в крови определяется выраженная нормохромная анемия. Уровень гемоглобина снижается до 30 г/л и ниже, наблюдаются ретикулоцитоз, нормоцитоз. При микроскопии эритроцитов отмечается наличие в них телец Гейнца (комочков денатурированного гемоглобина). При выраженном кризе отмечается выраженный сдвиг лейкоцитарной формулы влево вплоть до юных форм. В костном мозге выявляется гиперплазированный эритроидный росток с явлениями эритрофагоцитоза.
ДИАГНОЗ. Диагноз ставят на основании характерной клинико-гематологической картины острого внутрисосудистого гемолиза, лабораторных данных, выявляющих снижение ферментативной активности Г-6-ФДГ, и выявления связи заболевания с приемом гемолитических агентов.
ЛЕЧЕНИЕ. Прежде всего следует отменить препарат, вызвавший гемолиз. При нетяжелом гемолитическом кризе назначают антиоксиданты, применяют средства, способствующие увеличению глутатиона в эритроцитах (ксилит, рибофлавин). Одновременно дается фенобарбитал в течение 10 дней.
При тяжелом течении с выраженными признаками гемолиза необходима профилактика острой почечной недостаточности: проводится инфузионная терапия и гемотрансфузия. Применяют средства улучшающие почечный кровоток (эуфиллин в/в), диуретики (маннитол). В случае ДВС-синдрома назначают гепаринизированную криоплазму. Спленэктомия при этом виде гемолитических анемий не применяется.
Гемоглобинопатии – это наследственно обусловленные аномалии синтеза гемоглобинов человека: они проявляются либо изменением первичной структуры, либо нарушением соотношения нормальных полипептидных цепей в молекуле гемоглобина. При этом всегда возникает поражение эритроцитов, протекающее чаще всего с синдром врожденной гемолитической анемии (серповидно-клеточная анемия, талассемия). В то же время встречаются многочисленные случаи латентного носительства аномального гемоглобина. Гемоглобинопатии являются наиболее распространенными моногенными наследственными заболеваниями у детей. По данным ВОЗ (1983) на земном шаре насчитывается около 240 млн. человек, страдающих как структурными (качественными), так и количественными (талассемии) гемоглобинопатиями. Ежегодно в мире рождаются и умирают 200 тыс. больных людей. Значительная распространенность гемоглобинопатий в Закавказье, Средней Азии, Дагестане, Молдавии, Башкирии. Известно, что в норме гемоглобин взрослого человека состоит из нескольких фракций: гемоглобин А, образующий основную массу, гемоглобин F, составляющий 0,1—2%, гемоглобин А 2—2,5 %.
Это гетерогенная группа наследственно обусловленных гипохромных анемий имеющих различную тяжесть течения, в основе которых лежит нарушение структуры цепей глобина. У части больных основной генетический дефект заключается в том, что в клетках функционирует аномальная тРНК, тогда как у других больных наблюдается делеция генетического материала. Во всех случаях происходит снижение синтеза полипептидных цепей гемоглобина. Разнообразные типы талассемий с различными клиническими и биохимическими проявлениями связаны с дефектом любой полипептидной цепи. В отличие от гемоглобинопатии при талассемиях отсутствуют нарушения в химической структуре гемоглобина, но имеются искажения количественных соотношений гемоглобина А, гемоглобина F. Изменения структуры гемоглобина препятствуют нормальному течению метаболических процессов в эритроците, последний оказывается функционально неполноценным и разрушается в клетках ретикулоэндотелиальной системы. При талассемии в эритроцитах уменьшается содержание НЬА. В зависимости от степени снижения синтеза той или иной полипептидной цепи молекулы гемоглобина различают два основных типа талассемий: а и b.
источник
Наследственный сфероцитоз (НС) — группа патологических состояний, характеризующихся гемолитической анемией, сплело- мегалией и наличием эритроцитов сферической формы в периферической крови.
Кроме названия «наследственный сфероцитоз» или «микро- сфероцитоз», что менее точно, так как объем эритроцитов обычно в норме, данная нозология обозначается также как болезнь Мин- ковского-Шоффара (по фамилиям авторов, подробно описавших данную патологию в начале 1900-х гг.) или как-врожденная гемолитическая анемия.
Этиопатогенез. Заболевание врожденного, обычно наследственного характера. Обусловлено мутациями в генах, кодирующими мембранные белки цитоскелета эритроцитов. Наследуется, как правило, аутосомно-доминантно, однако не исключается и аутосомно- рецессивное наследование. Считается, что для первого типа наследования более характерна анемия легкой и средней степени тяжести, в то время как при рецессивном типе чаще встречается клинически тяжелая форма (Шиффман Ф. Д., 2000). Тип аномалии в цитоскелетных белках эритроцитов может быть различным: чаще встречаются аномалии спектрина и анкирина, реже отмечаются аномалии сегмента 3. и протеина 4.1 или 4.2.
При наследственном сфероцитозе имеется дефицит спектрина почти у всех больных и степень дефицита прямо коррелирует с тяжестью гемолиза. Кроме того, аномальные молекулы спектрина более чувствительны к иротеолизу с разрывом пептидных связей в местах локализации дефектов в полипептидных цепях. У одних больных имеется первичный дефект спектрина, в то время как у других выявляется дефект таких мембранных протеинов, как анкирин или белок полосы 3., которые прикрепляют снект- рин к мембране. Липидная структура мембраны при этом дестабилизируется, ведя к изменению площади поверхности и сферо- цитарной форме эритроцитов, теряется способность эритроцитов к деформации, нарушается работа Ка+/К4-насоса мембраны. В измененных эритроцитах может быть выявлено пониженное содержание солей калия, АТФ, ряда ферментов и другие биохимические нарушения. Такие ригидные клетки задерживаются и разрушаются в селезенке (Сторожок С. А. и др., 1997; Вуд М., Банн П., 2001 и др.).
Показано, что сфероцитарную форму основная масса эритроцитов с аномалиями мембранных белков приобретает при прохождении через селезенку, причем при прохождении клеток через синусы селезенки может нарушаться целостность мембраны эритроцита (Гусева С. А. и др., 2001). Преждевременная (не по мере старения) сферуляция эритроцитов в периферической крови ведет к укорочению продолжительности жизни эритроцитов и их разрушению макрофагами селезенки. Длительность жизни эритроцитов укорачивается до 12-14 дней, что требует компенсаторного усиления работы эритроидного ростка костного мозга (Шиффман Ф. Д., 2000; Гусева С. А. и др. 2001).
Клиническая картина. Первые признаки заболевания могут отмечаться как в детском, так и в зрелом возрасте. Иногда болезнь протекает скрыто и является случайной находкой, в том числе и у лиц пожилого возраста. Основным проявлением наследственной сфероцитарноп анемии является умеренно выраженная желтушная окраска кожи и видимых слизистых оболочек, нередко без клинически значимых проявлений анемии и интоксикации. К данной категории больных относится известное выражение о том, что они «более желтушны, чем больны». Отсутствие анемии длительное время у ряда больных объясняется компенсаторным повышением продукции эритропдных клеток в костном мозге. Такое компенсированное течение заболевания может наблюдаться у 25% больных (Гусева С. А. и др., 2001). Желтушная окраска кожи и склер нередко проявляется под воздействием таких провоцирующих факторов, как переохлаждение, беременность, повышенные физические нагрузки или эмоциональный стресс. Усиление гемолиза обычно наблюдается на фоне инфекционных заболеваний. У пациентов молодого возраста может отмечаться умеренное отставание в физическом развитии. В дальнейшем в клинической картине заболевания на первый план может выходить симптоматика, связанная с образованием пигментных камней и присоединением желчнокаменной болезни. Последняя выявляется примерно у половины пациентов (Гусева С. А. и др., 2001). По мере снижения компенсаторных возможностей кроветворения и развития анемии у больных появляются соответствующие жалобы и проявления анемического синдрома.
При осмотре больных, наряду с различной степени выраженности бледностью и иктеричностью кожных покрдвов и слизистых, определяется спленомегалия. Селезенка чаще плотная, безболезненная. Одновременно может выявляться и гепатомегалия, болезненность в проекции желчного пузыря.
Очень редко встречается осложнение НС в виде трофических язв голеней. Могут отмечаться цитоскелетные аномалии (высоко стоящее «готическое» небо, «башенный» череп, укорочение мизинцев, полидактилия, изменение расположения зубов), нередко сопровождающие наследственную сфероцитарную анемию. Наличие указанных аномалий у больных НС связывают как с вероятны-
10 Гематология. Нов. справочник ми хромосомными аномалиями, так и с компенсаторным расширением плацдарма кроветворения в условиях хронического гемолиза в период роста костей.
Таким образом, кроме анемии заболевание может проявлять себя следующими симптомами и осложнениями:
1) задержка роста и развития;
2) сниженная толерантность к нагрузкам;
Гемолитические кризы при НС у разных больных встречаются с различной частотой: у некоторых пациентов могут не наблюдаться на протяжении всей жизни, у других же появляются неоднократно каждый год. Такие кризы зачастую провоцируются инфекцией и сопровождаются слабостью, лихорадкой, желтухой, нередко — рвотой и болями в животе, что напоминает картину инфекционного гепатита или других инфекционно-воспалительных заболеваний, и в этом случае требуется проведение дифференциально-диагностических исследований.
У больных с хронической гемолитической анемией может развиться апластический криз, причину которого связывают с дефицитом микроэлементов и витаминов.
Диагностика. Диагноз сфероцитарной анемии основывается на наличии у пациента характерных морфологических изменений эритроцитов.
Из лабораторных /данных обращает на себя внимание наличие микроцитоза по мазку крови с отсутствием центрального просвета в эритроцитах. Средний диаметр эритроцитов, как правило, составляет 5,8-6,4 мкм при норме 7,2-7,5 мкм. При этом средний объем эритроцитов (МСУ) соответствует норме, а толщина клеток увеличена до 2,5-3 мкм (в норме 1,9-2,1 мкм). Соотношение между диам тром и толщиной эритроцитов («индекс сферичности»), в норме составляющее 3,4-3,9, при НС значительно снижается, причем количество сфероцитов при НС различается у отдельных больных: от относительно небольшой доли у одних до абсолютного большинства эритроцитов сферической формы у других. Выраженность признаков гемолиза в значительной степени зависит от общего количества микросфероцитов в циркуляции (Идель- сонИ.Л. и др., 1975).
Показатели насыщения эритроцитов гемоглобином (ЦП, МСН, МСНС) и уровень сывороточного железа обычно в норме, за исключением тех случаев, когда на фоне длительно существующего гемолиза в организме развивается железодефицитное состояние. Показатель МСНС у части больных может быть повышен.
Количество лейкоцитов и тромбоцитов обычно в пределах нормы. Появление нейтрофильного лейкоцитоза возможно в период гемолитического криза. В случаях апластических кризов отмечается резкое снижение уровня гемоглобина и эритроцитов, ретику- лоцитопения.
В костном мозге выявляется компенсаторное усиление’эритро- поэза в виде эритроидной реакции нормобластического типа с повышенным содержанием базофнльных, полихроматофильных, ок- сифильных эритрокариоцитов (нормобластов). Как и при других гемолитических анемиях, изменяется соотношение лейко : эритро (общего числа клеток лейкоцитарного и эритроидного рядов). В норме данное соотношение составляет в среднем 3:1, при с феро- цитарной же анемии оно может увеличиться до 1 : 1 и более. 3 некоторых случаях, особенно после гемолитических кризов, в мие- лограмме отмечается появление небольшого количества мегало- бластов, по-видимому, как отражение повышенного расхода фолиевой кислоты в период криза.
Отмечаются признаки гемолиза, такие как увеличение уровня ретикулоцитов, повышение содержания билирубина различной степени выраженности за счет непрямой фракции и повышение уровня уробилина в моче. Осмотическая резистентность эритроцитов у большинства больных снижена: при норме 4,6-4,8 г/л — для начального гемолиза и 3,2-3,3 г/л — для полного гемолиза у больных НС показатели могут быть увеличены до 5-6 и 3,8-4 г/л соответственно. Гемолиз начинается при концентрации солей, близкой к физиологическому раствору. Более показательно у данной группы больных определение осмотической резистентности после суточной инкубации эритроцитов и исследование кислотных эритрограмм с определением скорости распада эритроцитов при рН 3,0, отражающие повышенную хрупкость эритроцитов (Идель- сон И. Л. и др., 1975).
При исследовании белков мембраны эритроцитов по специальным методикам электрофореза в полиакриламидном геле можно идентифицировать дефекты отдельных мембранных белков (Сторожок С. А. и др., 1997).
Дифференциальный диагноз проводится с желтухами другой этиологии (инфекционным гепатитом, обструктйвной желтухой, синдромом Жильбера и другими), иммунной гемолитической анемией, спленомегалиями другой этиологии. При дифференциальной диагностике наряду с выявлением морфологически измененных эритроцитов, отрицательной пробой Кумбса и другими лабораторными данными немаловажное значение могут иметь тщательно собранный семейный анамнез и обследование родственников больного для выявления у них признаков НС.
Лечение. При клинически компенсированном состоянии больного, отсутствии значимого гемолиза и анемии терапия обычно ограничивается симптоматическими средствами, в том числе направленными на профилактику развития желчнокаменной болезни (желчегонные, фитотерапия и рациональная диета).
При тяжелом гемолизе с выраженной анемией и при апласти- ческих кризах с низким уровнем гемоглобина производятся трансфузии эритроцитарной массы.
Как и при других формах гемолитических анемий, у больных НС нередко развивается дефицит фолатов, в связи с чем в лечении этой категории больных используется фолиевая кислота.
Одним из основных методов терапии у больных сфероцитарной анемией является спленэктомия. Оперативное лечение считается показанным больным с наличием клинических проявлений гемолитической анемии средней и тяжелой степени или ее осложнений, в том числе при наличии желчнокаменной болезни, особенно у лиц молодого возраста. Детям младшего возраста спленэктомия производится редко из-за высокого риска тяжелых постспленэктомиче- ских инфекционных осложнений. Многие авторы рекомендуют при подготовке к операции проведение больным иммунизации поливалентной вакциной против пневмококков, Наето/Низ т/!иещае и менингококков за несколько недель до спленэктомии (Вуд М., Банн П., 2000; Гусева С. А. и др., 2001). В результате удаления селезенки прекращается или значительно уменьшается гемолиз эритроцитов и увеличивается продолжительность их жизни, причем этот эффект наиболее выражен у пациентов с дефектом спектрина и анкирина (КеИепе К. е! а1., 2002). Проведение спленэктомии целесообразно сочетать с холецистэктомией, особенно у пациентов с признаками холестаза и проявлениями желчнокаменной болезни.
источник