Беременность и роды у женщины с синдромом Марфана и акромегалией
М.Г. Газазян, И.С. Лунева, В.М. Саруханов,
О.П. Дубогрызова, О.Я. Янчук.
Курский государственный медицинский университет.
Кафедра акушерства и гинекологии
(зав. кафедрой д.м.н., профессор М. Г. Газазян).
Перинатальный центр Курского городского роддома
(заведующий В. М. Саруханов).
Для многих специалистов будет интересен впечатляющий по степени соматической тяжести и редкости, а также по подходу к родоразрешению и анестезиологическому обеспечению случай беременности и родов у женщины с синдромом Марфана и признаками акромегалии. По нашему мнению он представляет академический и практический интерес для анестезиологов (интенсивистов), акушеров — гинекологов, генетиков и эндокринологов.
Синдром Марфана (СМ) относится к наследственным доминантным болезням соединительной ткани с высокой пенентрантностью мутантного гена. Впервые описанная Вильямсом в 1876г, в последующие годы эта болезнь наблюдалась Марфаном (1896), давшим патологии свое имя. При СМ мутация в гене, ответственном за синтез белка соединительно-тканных волокон фибриллина, приводит к блоку его синтеза, соответственно соединительная ткань обладает повышенной растяжимостью. Симптоматика СМ многосистемная и разнообразная. Клинический по-лимофизм по степени выраженности симптомов очень многообразен. Частота СМ в популяции равна 1:10000-1:15000. Популяционных и этнических различий в частоте и клинической картине болезни не отмечено [4]. Диагноз СМ ставится при условии обнаружении у пациента нарушений скелета с вовлечением в патологический процесс двух других систем, включая одну с наиболее специфическими проявлениями (глаза, скелет, сердце).
Изучение патогенетических механизмов нарушения роста при СМ рядом авторов (Семячкина А.Н., Семерова Н.Б., Любченко Л.Н., 1983) позволило установить вовлечение в патологический процесс органов эндокринной системы, в первую очередь соматотропной функции гипофиза и инсулярного аппарата поджелудочной железы. Обнаружены высокие показатели соматотропина (в 5-10 раз превышающие показатели нормы) и парадоксальная реакция гормона на глюкозо-толерантный тест у пациентов с тяжелыми клиническими проявлениями СМ. По видимому эндокринные расстройства, не только усугубляют тяжесть заболевания, но в ряде случаев являются одним из основных патогенетических механизмов формирования кардинальных симптомов болезни [6]. Одним из ведущих клинических проявлений в представленном нами случае, является гигантизм и акромегалия относящиеся к нейроэндокринным заболеваниям, в основе которых лежит патологическое повышение ростовой активности. Патологическим считается рост у женщин свыше 190 см. После окостенения эпифизарных хрящей гигантизм переходит в акромегалию. Ведущим признаком акромегалии является рост тела, но не в длину, а в ширину, что проявляется в диспропорциональном периостальном увеличении костей скелета и внутренних органов, которые сочетаются с характерными нарушениями обмена веществ [5].
Необходимо остановится на вопросах вынашивания беременности и родо-разрешения у женщин с СМ. По данному вопросу крайне мало литературных сведений. R-F.Pyerirz (1981) на основании собственных наблюдений, проанализировал осложнения при беременности у лиц с СМ. Автор подчеркнул главенствующее значение состояния сердечно-сосудистой системы при решении вопроса о возможности вынашивания беременности [6]. Беременность у женщин с синдромом Марфана и акромегалией наступает крайне редко. В литературе имеется описание четырех случаев беременности у женщин с акромегалией [2,7]. В литературе мы не встретили описания сочетания беременности с синдромом Марфана с выраженными признаками акромегалии.
Именно клиническая редкость сочетания данной патологии с беременностью явились основанием поделится с коллегами особенностями родоразрешения и анестезиологического ведения пациентки М., 38 лет, поступившей в клинику на сроке беременности 36 недель.
Жалобы при поступлении на слабость, одышку в покое, отеки нижних конечностей, онемение в руках, ограничение движения в суставах. Пациентка страдает синдромом Марфана, по поводу заболевания состоит на учете в медико-генетической консультации, инвалид 1 группы (инвалид детства). Женщина живет в деревне с родителями, которые за ней ухаживают, выполняет несложную работу по дому. Закончила 10 классов общеобразовательной школы. Со слов выяснено, что в детстве отличалась от сверстников высоким ростом, с 12 — 13 лет началось искривление позвоночника. Часто меняла обувь и одежду, т.к. размер ноги быстро увеличивался. В настоящее время размер обуви — 44 (28 см).
Менструальная функция с 13 лет, не регулярная, когда была последняя менструация, пациентка не помнит. В браке не состоит. Целенаправленного, углубленного обследования и лечения не проводилось.
Состояние при поступлении тяжелое. Сознание ясное. Интеллект сохранен. Женщина высокого роста -178 см, без учета кифосколиотического искривления позвоночника. Кифосколиоз 4 ст, килевидная грудина. Грудная клетка “сидит” на широких гребнях крыльев подвздошных костей (рис. 1, 2, 3).
Рис. 1-3. Для просмотра в увеличенном виде нажмите на соответствующий слайд
Вес 86 кг. Обращают внимание длинные большие руки, кисти, стопы. Лицо большое, но черты лица тонкие — заостренный нос, “птичий профиль”, узкие губы. Пальпируется увеличенная щитовидная железа (2 ст.) . Язык увеличен, “готическое небо”. Кожные покровы бледные, влажные, жирные. Мышечная ткань выражена на конечностях и практически отсутствует на ягодицах. На нижних конечностях — отеки. В легких дыхание везикулярное, ослабленное, хрипов нет, частота дыханий (чд) 36 в мин., разговорная одышка, АД 160 и 90 мм рт. ст. , 150 и 90 мм рт. ст. (Dex, Sin). Границы сердца смещены вправо, отмечается смещение границы относительной тупости вправо на 5 см от правого края грудины. При аускультации тоны сердца приглушены, не ритмичные, чсс 96-106 уд в мин., экстрасистолия . Проба с задержкой дыхания 9 секунд. Живот увеличен в поперечном размере, мягкий при пальпации , на уровне пупка определяется верхний полюс матки. Печень из под реберной дуги не пальпируется.
В матке определяется плод, находящийся в поперечном положении, головка справа. Высота стояния дна матки над лоном 24 см, окружность живота 94 см. Размеры таза dist. spinarum 35 см, dist.cristarum 37 см, dist.trochanterica 40 см, con.extema 22 см. С/б плода выслушивается ритмичное 128-140 уд в мин. Срок последней менструации, и первое шевеление плода пациентка не помнит, в женской консультации не наблюдалось. При влагалищном исследовании выявлено, что наружные и внутренние половые органы сформированы правильно, малый таз емкий, широкий. Родовые пути к родам не готовы.
Ультразвуковое исследование матки — положение плода поперечное, бипа-риетальный размер головки — 88 мм, длина бедра — 68 мм. Плацента по передней стенке 2 — 3 ст. зрелости. Относительное маловодие. Сердцебиение плода ритмичное 136 уд в мин. Дыхательные движения и активность плода снижены. Заключение. Беременность 36 недель. Относительное маловодие. Биофизический профиль плода 6 — 7 баллов. Предполагаемый вес плода 3000,0 — 3100,0.
Ан. крови общий: НВ 96 г/л. Ц.п. 0,85 Эр. 3,1х10 Тромбоциты 180х10, z 9,7х10 Соэ 2 мм/час, Ht — 38 %. В моче белок 0,095 г/л. Z 5-8 п/зр. Уд. вес 1009.
Биохимический анализ крови и показатели коагулограммы в пределах физиологической нормы беременных.
Электрокардиограмма (ЭКГ).
Эпизодически параксизмальная мерцательная аритмия, тахисистолическая форма. На ряде ЭКГ частые политопные экстрасистолы. Гипертрофия правого предсердия. Блокада правой ножки пучка Гиса (возможно как эквивалент гипертрофии правого желудочка). Гипертрофия левого желудочка.
Ультразвуковое исследование сердца (четкая визуализация из-за выраженной деформации грудной клетки затруднена). Аорта не уплотнена, не расширена, на уровне фиброзного кольца — 3 см. Полость левого, желудочка — 4,6 см (не расширена). МЖП не утолщена (1,1 см); задняя стенка левого желудочка не утолщена (0,9 см). Аортальный клапан не утолщен, кальциноза нет. Митральный клапан- проти-вофаза есть, створки не утолщены, кальциноза нет. Правый желудочек расширен до 4,3 см, стенка утолщена до 0,5 см. Правое предсердие увеличено — 5,2х3,0 см. Среднюю длину аорты определить не удается . Заключение Гипертрофия и дилятация правого желудочка. Дилятация правого предсердья. Пролапс трикуспидального клапана 2 ст.
Рентгенограмма черепа в боковой проекции (рис.4). Форма черепа долихо-цефалическая. Сагитальный размер черепа 25 см. Кости свода утолщены , уплотнены, отмечается гипертрофия затылочного бугра. Турецкое седло больших размеров, круглое. Вертикальный размер турецкого седла — 2 см, сагитальный размер — 2 см. Индекс «седла» равен 1 (H/S). Индекс «седло-череп» (Martinez- Farinas)
Контуры турецкого седла истончены, отмечается частичный остеопороз спинки турецкого седла, прерывистость его контуров. Сосудистый рисунок прослеживается слабо.
Заключение: Большие размеры турецкого седла, “индекс седла” равный 1 и индекс “седло — череп” выше 7,3 , говорит об объемном образовании гипофиза с разрушением стенок турецкого седла.
Консультация окулиста. Среды прозрачные, подвывиха хрусталика нет. Глазное дно: диски розовые, границы четкие, сосуды сетчатки извитые, вены расширены. Заключение : Ангиопатия сетчатки.
источник
Добрый день, друзья. Рад снова видеть Вас на страницах сайта для высоких людей!
Акромегалия (от греч. akron конечность и megas, megalu огромный) — эндокринная патология, характеризующаяся нарушением функции передней доли гипофиза и увеличением выработки соматотропного гормона роста.
Основным симптомом заболевания является непропорциональное увеличение отдельных частей тела человека. Симптомы болезни проявляются только у взрослых людей после завершения роста организма, спустя 5-7 лет после начала течения патологии. Поэтому выявить акромегалию на ранних стадиях возможно лишь при помощи лабораторных исследований уровня соматотропного гормона. У детей акромегалия проявляется лишь в виде увеличения роста.
- Высокий рост
- Укрупнение черт лица (скуловых костей, челюсти, надбровных дуг, мягких тканей)
- Увеличение грудной клетки
- Непропорциональное увеличение (утолщение) размеров стоп и кистей рук.
- Боль в области суставов
- Головная боль
- Артериальная гипертония
- Расстройство половой функции у мужчин и менструального цикла у женщин
Избыточная выработка гормона роста может способствовать видимой деформацию внешности больного: нижняя челюсть становится массивной, надбровные дуги более выраженными, увеличивается в размерах нос. Нередко отмечается макроглоссия — увеличение в размерах языка, что ведет к изменению прикуса. Гипертрофируется гортань и голосовые связки, в связи с чем меняется и голос. Он становится низким и хрипловатым, напоминает голос курильщика с многолетним стажем. Изменения происходят не сразу, очень часто больной и его близкие родственники их не замечают.
Для акромегалии свойственна деформация скелета: грудная клетка приобретает бочкообразную форму, межреберные промежутки становятся намного шире, а позвоночник искривляется настолько, что у больного ограничивается функциональность опорно-двигательного аппарата. Хрящевая ткань суставов начинает разрушаться, что чревато артрозами и артритом.
Акромегалия способствует избыточному выделению кожного сала и стимуляции функций потовых желез. Помимо видимых внешних проявлений наблюдаются и внутренние:
- Увеличивается в размерах сердце
- Заметно увеличиваются печень и почки
Такие симптомы могут послужить причиной дисфункции внутренних органов.
Больные акромегалией отмечают повышенную утомляемость и постоянную сонливость. В клинических условиях у них можно диагностировать сердечную недостаточность и артериальную гипертензию. 90% носителей недуга подвержены возникновению синдрома сонных апноэ (расстройство дыхательной функции, во время которого наблюдается частая остановка дыхания во время сна).
Болезнь ограничивает половую функцию. У женщин наблюдается избыток пролактина, в связи с чем менструальный цикл становится нерегулярным. Помимо этого они сталкиваются с рядом трудностей в плане осуществления репродуктивных способностей. Что касается мужчин, то у многих уже в молодом возрасте снижается потенция.
По мере разрастания опухоли передней части гипофиза у больных повышается внутричерепное давление. Они становятся раздражительными и светочувствительными. Снижается обонятельная, тактильная и визуальная восприимчивость.
Пациенты, страдающие акромегалией, больше чем другие подвержены заболеваниям эндокринной системы. Высок риск развития опухолей щитовидной железы, матки, а также органов пищеварительной системы.
О данном заболевании мало что известно. Ранее его путали с гигантизмом, но у людей, страдающих гигантизмом, симптомы проявляются с юношеского возраста, а у больных акромегалией – только после взросления.
Самый известный больной акромегалией в истории – французский рестлер Морис Тийе. В 40-х годах Морис стал одним из самых знаменитых борцов. Также он дважды был признан Американской Ассоциацией Рестлеров чемпионом мира в своей области в категории тяжеловесов. За эти заслуги преданные фанаты Тийе прозвали его «Французским Ангелом».
Всю свою юность Морис мечтал об успешной карьере юриста, но она не сложилась из-за прогрессирования болезни. Известно, что талантливый спортсмен с юмором относился к своему недугу. Однажды в интервью для газеты Lowell Sun Newspaper он сказал следующее: «Может, с таким лицом я и смог бы стать адвокатом, но мой голос, похожий на ослиный рев, просто невозможно слушать, поэтому я пошел в ВМС».
Интересный факт про Мориса Тийе. Именно французский спортсмен стал прототипом всеми любимого персонажа мультфильма Шрек. Незабываемая внешность и харизма спортсмена были полностью скопированы художниками.
Самоирония наблюдалась во многих поступках Мориса. В один прекрасный день Морис прогуливался по коридорам палеонтологического музея, где не упустил возможности с удовольствием позировать для фотокамер прессы рядом с восковыми фигурами неандертальцев. Как признавался сам Тийе — его очень забавляло их внешнее сходство.
При лечении акромегалии врачи стараются добиться ремиссии заболевания путем нормализации секреции гормона роста (соматотропина).
В современной эндокринологии применяются 4 способа устранения недуга:
- Медикаментозный
- Хирургический
- Лучевой
- Комбинированный
Для нормализации уровня соматотропина назначаются лекарства, подавляющие его секрецию. Людям с противопоказаниями к приему гормональных препаратов рекомендуется лучевая терапия, во время которой происходит направленное воздействие на гипофиз гамма-излучением.
Самым эффективным методом лечения является хирургическая операция по удалению опухоли передней части гипофиза. Согласно статистике, у 70% пациентов наблюдается стойкая ремиссия, а 30% заболевших полностью выздоравливают.
Что касается профилактики заболевания, то четких рекомендаций нет. Следует
- Избегать черепно-мозговых травм
- Вовремя лечить инфекции, поражающие носоглотку
При выявлении признаков Акромегалии следует немедленно обратиться к эндокринологу за консультацией.
Болезнь роста. Два простых теста как выявить акромегалию:
Болезни, меняющие внешность. Программа здоровье с Еленой Малышевой. Диагностика и лечение болезни:
1. Причины высокого роста — статья, в которой рассмотрены основные причины высокого роста человека.
2. Акромегалия в википедии — об Акромегалии в свободной энциклопедии.
4. Акромегалия на здоровье@mail.ru — Статья о болезни. Причины появления, прогноз лечения
5. И.И.Дедов,Г.А.Мельниченко, В.В.Фадеев., Эндокринология учебник «ГООЭТАР-Медиа».2012.
Понравилась статья? Расскажи друзьям:
источник
Высказывались версии, что такие большие, широкие кисти рук могут быть симптомом болезни. Например, синдром Марфана характеризуется разрастанием конечностей, увеличением хрящей, связок и суставных капсул. Но у Рахманинова ничего такого не было. Руки его были хоть и огромные, но красивые и изящные.
О великом композиторе Генделе рассказывают вот что: приехав однажды в голландский Харлем, он отправился в местный собор, что славился своим органом. Получив разрешение ознакомиться со знаменитым инструментом, он заиграл… Местный органист, сам искуснейший музыкант, войдя в собор, замер в изумлении. Такой игры он никогда не слыхал! «Кто это играет? – воскликнул он. – Если ты не ангел и не дьявол, то, наверное, Гендель!» Когда он узнал, что это и вправду Гендель, то изумился ещё больше. «Как вы это сделали? – воскликнул он. – Нет на свете человека, который смог бы сыграть те пассажи, что играли вы, своими десятью пальцами. Человеческие руки просто не могут этого!» «Знаю, – спокойно проговорил Гендель, – некоторые ноты мне пришлось брать кончиком носа. »
А герою нашей стати вряд ли пришлось бы носом жать на клавиши. О волшебных руках Сергея Рахманинова ходит множество легенд, и они по праву занимают своё место в книге рекордов Гинесса. Известно, что кисти его обладали самым большим охватом клавиш – дюжина! При этом левой Рахманинов легко брал аккорд до ми-бемоль соль до соль! И его поистине огромные руки были дивно красивы: цвета слоновой кости, гладкие, без вздувшихся вен, узлов на пальцах и прочих дефектов, которыми обычно поражены руки постоянно концертирующих пианистов. Во всех его биографиях рассказывается о том, что он любил носить туфли на кнопках. И чтобы старенький композитор не повредил пальцы, эти кнопки трогательно застегивала его супруга. Однажды, разозлённый назойливым фотографом-папарацци, он вскинул руки, закрывая лицо от камеры. Но это не спасло композитора – фотография рук появилась в газетах с подписью «Эти руки стоят миллион долларов».
Ну а что уж говорить о знаменитом концерте №2 для фортепиано, который заслуженно считают истинным шедевром творчества Рахманинова! Произведение держит одно из первых мест по популярности, что говорит о его яркости и совершенстве. Многие знают, что концерт этот посвящён русскому врачу Николаю Владимировичу Далю. Как-то в 1928 году в Бейруте на концерте приезжего солиста-пианиста, которому аккомпанировал местный любительский оркестр американского университета, произошел интересный случай. Давали 2-й фортепианный концерт Рахманинова. Всё прошло блестяще, полный триумф, овация. Но тут пианист направляется в оркестр, вытаскивает из группы альтистов немолодого человека с донкихотской эспаньолкой, который смущенно и неловко кланяется публике. Этот альтист-любитель – известный врач-невролог Николай Владимирович Даль, в своё время учившийся в Париже у знаменитого доктора Шарко. Именно ему посвятил Рахманинов своё гениальное произведение.
В середине марта 1897 года молодой Рахманинов испытал тяжкий удар – провал своей 13-й симфонии, на которую он возлагал большие надежды. Отзывы были разгромными. Цезарь Кюи, музыкальный критик и композитор, заявил, что, мол, обитатели преисподней пришли бы в экстаз от симфонии господина Рахманинова. Рахманинов погрузился в отчаяние. Он бродил по ночному городу, лишившись сна и аппетита, всё более склоняясь к самоубийству. Он поклялся не писать музыки вовсе. К тому же девушка, за которой он ухаживал и которую любил, вышла замуж за другого. Депрессия и тревожное состояние захватили молодого композитора на целых три года. Он жаловался на сильные боли в спине и конечностях, тяжкую усталость, бессонницу. Кроме того, знаменитый в Москве профессор-терапевт Остроумов заявил родным Рахманинова, что есть все основания опасаться за его рассудок…
Портрет Сергея Рахманинова. 1925 г. Константин Сомов
Приятель композитора, доктор Грауэрман, под предлогом прогулки привел Рахманинова к Далю. С того дня композитор почти полгода посещал врача. Но Даль не называл эти беседы лечением. Рахманинов приходил к Далю поговорить, послушать музыку, пообщаться. Тогдашние русские медики были весьма тесно связаны с искусством, знали толк в живописи и скульптуре, были заядлыми театралами, играли в любительских спектаклях, успешно музицировали. Таков был и Даль, превосходно игравший на альте и виолончели, не чуравшийся любительской сцены.
Даль лечил Рахманинова исподволь, потихоньку, слово за словом открывая Рахманинову суть его болезни. Эти разговоры снимали стресс и нервное напряжение, отвлекали от печальных раздумий о творческом крахе. Наделенный неистощимой фантазией и пылким воображением, композитор, как и все подобные личности, был весьма восприимчив к гипнозу, который Даль весьма успешно использовал.
Постепенно к Рахманинову возвращались как вера в свои силы, так и энергия жизни и творчества. Свой сверкающий солнечный концерт №2 Рахманинов создал весной 1901 года – сразу же, как преодолел болезнь. Прямо на титульном листе оригинальной партитуры он написал посвящение Далю, который был на премьере. «Это не мой, это Ваш концерт», – сказал он врачу. И с тех пор Николай Владимирович по просьбе композитора часто сидел в самом первом ряду партера на его выступлениях. Но не затем, чтобы демонстративно и безнаказанно есть лимоны, как могут подумать многие читатели, а напротив, чтобы поспособствовать концентрации на творческом процессе, помочь композитору справиться с волнением на эстраде под взглядами многочисленной публики.
Рахманинов всегда считал, что Даль действительно спас его, а не просто излечил. Об этом он писал многим своим корреспондентам. С точки зрения современной медицины такое лечение не только возможно, но и нередко применяется при развитии подобных заболеваний. И в те времена неврологи вполне могли заниматься и занимались тем, что сегодня мы называем психотерапией. Именно психотерапия может стать основой для борьбы с депрессией. Когнитивно-поведенческая терапия, как считает современная врачебная наука, вполне эффективна в таких случаях. Грамотный психотерапевт мог сам справиться с недугом, тем более что в те времена нормальные фармацевтические препараты практически отсутствовали. Больных в огромных дозах потчевали кофеином. Использовали и настойку каннабиса, и даже кокаин. Что же касается гипнотерапии, то в России она исторически признана одним из методов лечения расстройств, связанных с эмоциональной нестабильностью. Но на Западе в настоящее время именно когнитивно-поведенческая терапия является мейнстримом. Гипнотерапию там считают необходимой лишь при легких неврозах, а тяжёлые расстройства лечатся другими методами.
Вернемся к феномену рук Рахманинова, обсуждение которого не раз проводилось в медицинских кругах. Высказывались версии, что такие большие, широкие кисти рук могут быть симптомом болезни. Например, синдром Марфана или различные дисплазии соединительной ткани характеризуются разрастанием конечностей, увеличением хрящей, связок и суставных капсул. При синдроме Марфана наблюдаются «паучьи лапы» — арахнодактилия и гипермобильность (избыточная гибкость) суставов. Но у Рахманинова ничего такого не было. Руки его были хоть и огромные, но красивые и изящные, как указывалось выше.
Некоторые медики пытаются притянуть сюда и акромегалию, при которой наблюдается избыток гормона роста. Больные этим недугом обладают очень характерными приметами, заметными любому врачу-эндокринологу. При этой патологии характерна артериальная гипертензия, что у Рахманинова зафиксировано не было. В медицинской литературе рассказывается о связи акромегалии с депрессией. Очевидно, именно это и послужило поводом упомянуть её в связи с Рахманиновым.
Как известно, существует пока ещё не совсем понятная науке связь акромегалии и меланомы, от которой и погиб великий композитор. Рак кожи (меланома) был диагностирован у Рахманинова меньше чем за год до смерти. До самого последнего времени меланома считалась «королевой раковых опухолей», она сжигала людей очень быстро. Прорыва в ее лечении средствами иммунотерапии ученые добились только в середине 2010-х годов. Увы, в 40-е годы прошлого столетия ничего подобного не применялось. Мы можем лишь возрадоваться, что у Сергея Рахманинова всё-таки были эти почти 70 лет жизни для его блестящего творчества.
источник
Что такое синдром Марфана? Причины возникновения, диагностику и методы лечения разберем в статье доктора Боровикова О. И., гинеколога-эндокринолога со стажем в 10 лет.
Синдром Марфана (Marfan; СМ) — генетически обусловленное заболевание, при котором происходит системное поражение соединительной ткани. [1]
Этиологией заболевания является мутация в гене FBN1 (фибриллина 1), расположенном в коротком плече пятнадцатой хромосомы в локусе 21.1. [2]
Наследование заболевания происходит по аутосомно-доминантному типу, характеризуется высокой пенетрантностью (частотой появления гена) и различной экспрессивностью. [5]
Соотношение представителей мужского пола и женского одинаковое.
Наблюдается постоянно прогрессирующее развитие заболевания. У новорожденных детей выявляются удлинённые тонкие пальцы на верхних и нижних конечностях и удлинённые тонкие конечности (долихостеномелия). [1] У таких пациентов, помимо долихостеномелии, отмечается:
- повышенное физическое развитие;
- недостаток веса;
- удлинённый череп;
- вытянутое лицо;
- арахнодактилия (аномально удлинённые узкие пальцы);
- слабость и недоразвитие мышечной системы и жировой клетчатки;
- неловкие движения. [3]
Кожа имеет повышенную растяжимость, разболтанные суставы. У большинства больных наблюдается высокое аркообразное нёбо, изменения формы грудной клетки (воронкообразная, килевидная) и искривления позвоночника (сколиоз в 60%, кифоз (изгиб позвоночника с образованием горба), ювенильный остеохондроз), уплощение свода стопы, аускультативные признаки порока сердца (шумы). [4] Длина третьего пальца руки — 10 см и больше (скрининговый тест у детей 7-18 лет): возрастает соотношение размаха верхних конечностей к длине тела.
Офтальмологические симптомы (близорукость, подвывих хрусталика в 75% случаев, его округлость или гипоплазия, отслойка сетчатки) и астенические признаки (усталость, вялость) обращают на себя внимание со второго года жизни, изменения формы грудной клетки появляются в возрасте старше четырёх лет, патология сердца и сосудов выявляется в дошкольном возрасте. [1]
Почти у всех больных выявляются пороки сердца и аорты. Часты бедренные и паховые грыжи, поражение клапанов в венах, их варикозное расширение, геморрагический синдром, рецидивирующие вывихи, поражение лёгочной системы (самопроизвольный пневмоторакс, эмфизематозное расширение лёгких), опущение почек. [2]
В четверти случаев зарегистрировано снижение интеллекта, у половины пациентов выявляются нарушения эмоционально-волевой сферы. Часто появляются депрессивные состояния, нейроциркуляторная дистония. [3]
По данным многих исследований, абсолютное большинство больных с синдромом Марфана отмечают ухудшение эмоционального фона, утрату чувства радости и увлечённости профессиональной деятельностью, частую смену настроения, повышенную возбудимость, чувство тревоги. Результатом этого является снижение социальной активности, ухудшение качества жизни и значительное уменьшение социальной адаптации. [3]
У таких пациентов часто наблюдается трахеобронхиальная дискинезия (нарушение дыхательной системы) за счёт слабости соединительнотканного каркаса бронхов. Это проявляется рецидивирующими воспалительными заболеваниями бронхолегочной системы, обструктивными нарушениями, бронхиальной астмой, эмфиземой лёгких (повышенное содержание воздуха в лёгочной ткани). [4] Встречаются осложнения, которые проявляются скоплением воздуха в грудной клетке, сопровождающиеся сдавлением лёгких и средостения (срединной области грудной клетки), подкожной эмфиземой. Наблюдается неадекватный ответ на бронхолитики. Обструктивные явления (непроходимость) затрагивают преимущественно верхние отделы респираторного тракта. [3]
Описаны характерные изменения на электрокардиограмме, включающие синдром раннего возбуждения желудочков, преждевременные желудочковые комплексы, нестабильность конечной части желудочкового комплекса в задненижних отведениях. [3]
Патология ритма чаще всего проявляются блокадой правой ножки пучка Гиса или смешанной экстрасистолией. [6]
У больных синдромом Марфана с патологией ритма сердечной деятельности и проводимости синдром вегетативной дисфункции чаще протекает по ваготоническому типу, в виде пресинкопальных, обморочных и астеновегетативных состояний, болезненных ощущений в области сердца, цефалгии напряжения (головной боли) и зачастую сочетается с психопатологическими расстройствами. [4]
Органы пищеварения также задействованы в патологическом процессе, что проявляется дискинезией (нарушением моторики) билиарного тракта со снижением моторики гладкомышечной мускулатуры, недостаточностью кардии, грыжевыми выпячиваниями пищеводного отверстия диафрагмы, аномалиями желчевыводящих протоков, долихосигмой (увеличением сигмовидной кишки), хроническим гастродуоденитом (воспалением слизистой желудка и двенадцатиперстной кишки), дисбиозом (нарушением нормальной микрофлоры) кишечника, изменениями поджелудочной железы. [3]
У пациентов с синдромом Марфана чаще, чем у здоровых людей, встречаются приобретённые аномалии почек: повышенная подвижность почек, нефроптоз (опущение почки), пиелоэктазии (аномальное расширение лоханок), повышена частота удвоения почек.
Более половины веса человека представлено соединительной тканью, из неё состоит наша главная опора — скелет, внешние покровы — кожа. Сосуды, кровь и лимфа тоже состоят из соединительной ткани.
К клеткам соединительной ткани относятся фибробласты и их разновидности (остеобласты, хондроциты, одонтобласты, кератобласты), макрофаги (гистиоциты) и тучные клетки (лаброциты). [7]
Мезенхима — проводник конституциональных, генетических и эпигенетических составляющих жизни человека. Патология соединительной ткани детерминирует определенное патологическое действие на весь организм в целом, на его физиологию и его конституциональные особенности. [3]
При болезни Марфана происходит замена нуклеотидов в гене, содержащем информацию о структуре пептида фибриллина-1. Этот белок относится к гликопротеидам, принимает участие в микрофибриллярном комплексе, он обеспечивает основу эластических фибрилл соединительной ткани.
Межклеточный матрикс позволяет соединительной ткани поддерживать постоянную структуру, в нём находится огромное количество факторов роста, которые обеспечивают постоянное обновление клеток.
В крупных сосудах, связочном аппарате содержится большое количество эластиновых фибрилл, поражение которых и даёт основные клинические проявления синдрома Марфана.
При синдроме Марфана значительно поражается трансформирующий фактор роста бета (TGF-β), нарушается связывание его неактивной формы, что приводит к повышению биоактивности данного фактора, с чем связано появление многих проявлений болезни. [4]
Патология фибриллина приводит к патологии формирования волокон, что вызывает утерю прочности и эластичности кожи и других соединительнотканных структур.
Изменение структуры коллагеновых волокон приводит к нарушению первичного звена гемостаза у пациентов с синдромом Марфана. [6]
Имеются данные о дефектах мембранных и цитоплазматических механизмов проведения сигнала непосредственно в самом тромбоците, приводящих к нарушениям агрегации (объединения). Показано наличие самостоятельного мембранного дефекта тромбоцитов, протекающего с нарушением реакций высвобождения и транспорта внутриклеточного кальция. [6]
Эластические фибриллы имеют вполне определенные механизмы участия в системе гемостаза. В сосудах с низкой скоростью сдвига происходит адгезия («прилипание») тромбоцитов к эластину через фибронектин. [7] Регистрируется снижение его уровня в крови у людей с синдромом Марфана. Фибронектин, в свою очередь, образуется в клетках эндотелия и участвует в последующих репаративных процессах, создавая основу для производства других компонентов соединительной ткани — фибробластов. [4] Таким образом, совершенно неоспоримо участие сосудистой стенки в реакциях свертываемости крови, и неизбежен вывод о возможных патологиях протекания нормальных гемостатических процессов при изменении состояния её структурных компонентов и процессов сосудистой регуляции.
Отмечена роль гормонального дисбаланса в развитии и усугублении дефектов соединительнотканных структур. [3]
Тромботические проявления детерминированы нарушением реологии (вязкости) крови в патологически извитых сосудах брахиоцефальной зоны. [3]
Поражение желудочно-кишечного тракта детерминировано тем, что эта система богата коллагеном. Наблюдаются дискинезия билиарного тракта по гипомоторному типу, грыжи пищеводного отверстия диафрагмы, аномалии желчных путей, долихосигма, хронический гастродуоденит со стёртой клинической картиной, склонностью к торпидному течению. [3]
- стёртая (поражено не более двух систем, изменения выражены незначительно);
- выраженная (незначительные изменения в трёх системах либо значительное поражение одной и более систем).
Выделяют различные типы по степени тяжести:
Частота тяжёлых форм — 1 к 25000-50000 (при общей частоте диагностированных случаев 1 к 10000-15000).
- прогрессирующая форма;
- стабильная форма.
Чаще всего первые признаки синдрома Марфана проявляются еще в детском периоде, с возрастом происходит прогрессирование симптомов, усиление клинических проявлений.
К самым частым осложнениям синдрома Марфана относятся:
- Снижение зрения, вплоть до слепоты, обусловленное слабостью цинновой связки (ресничного пояска) и подвывихом, вывихом хрусталика. [7]
- Сердечная недостаточность по застойному типу, обусловленная нарушением сократимости сердечной мышцы, недостаточностью митрального клапана. [6]
- Разрывы крупных сосудов, связанные с дилатацией (расширением), истончением стенки сосудов. Чаще всего происходит поражение аорты (в основном из-за изменения гемодинамики при беременности). [7]
- Расслаивающая аневризма аорты, приводящая к смерти больных.
Диагностика Синдрома Марфана основывается на клинических данных, выявлении изменений в гене FBN1. [5]
Часто при сборе генеалогического анамнеза выявляются родственные случаи со скрытым течением заболевания. [1]
Способы обнаружения арахнодактилии: [3]
- Симптом Steinberg (признак первого пальца). Первый палец виден из-под hypothenar при напряжённом кулаке.
- Симптом Walker-Murdoch (признак запястья). При обхватывании кистью в области лучезапястного сочленения контралатеральной верхней конечности первый палец заходит за пятый.
- Определение пястного индекса. Определяется при помощи рентгенографии. Средняя длина пясти, делённая на усреднённую ширину отрезка от второй до четвертой пястной кости. При нормальном соотношении этот показатель соответствует 5,4-7,9, в то время, как при синдроме Марфана — больше 8,4.
В 2010 году группа специалистов систематизировала международные Гентские критерии для верификации синдрома Марфана. Верификация зависит от данных генеалогического анамнеза. [3]
При отсутствии генеалогического анамнеза:
- увеличение диаметра аорты >, = 2 ϭ + эктопия хрусталика = СМ;
- увеличение диаметра аорты >, = 2 ϭ + выявленные изменения в гене FBN1 = CM;
- увеличение диаметра аорты >, = 2 ϭ + >, = 7 системных признаков = СМ;
- эктопия хрусталика + наличие изменений в гене FBN1 + дилатация аорты = СМ;
При наличии генеалогического анамнеза:
- Эктопия хрусталика + случай СМ в семье = СМ;
- >, = 7 системных проявлений + случай СМ в семье = СМ;
- увеличение диаметра аорты >, = 2 ϭ + случай СМ в семье = СМ.
В пятнадцати процентах появление ребёнка с синдромом Марфана спорадическое (случайное), у родителей могут быть слабые проявления. У родственников пациентов встречаются заболевания желудочно-кишечного тракта, поражения позвоночника, заболевания глаз. [3]
При малейшем подозрении на синдром Марфана необходима консультация офтальмолога. В анализе мочи таких пациентов отмечается повышение уровня оксипролина, гликозаминогликанов, но эти показатели низкоспецифичны, могут быть при различных дисплазиях соединительной ткани. Выделение оксипролина является показателем тяжести заболевания. Наблюдается нарушение свертываемости крови на тромбоцитарном уровне. [3]
Оценка системных признаков вовлечённости соединительной ткани
источник

Болезнь (синдром) Марфана – это наследственная патология соединительной ткани. Поскольку соединительная ткань является основой различных органов человека, то и проявления заболевания весьма разнообразны. Самыми типичными являются аномалии сердечно-сосудистой системы, опорно-двигательного аппарата и глаз. Диагностика синдрома Марфана требует использования самых разных методов. Лечение включает в себя медикаментозные и оперативные способы, причем наблюдаться больной должен у нескольких врачей. Из этой статьи Вы сможете узнать о причинах, симптомах, диагностике и вариантах лечения данного заболевания.
Синдром Марфана формируется еще во внутриутробном периоде, ребенок рождается уже с имеющимися нарушениями. Это, пожалуй, наиболее часто встречающаяся наследственная патология соединительной ткани. Независимо от пола и расовой принадлежности, частота выявления болезни Марфана колеблется от 1: 5 000 до 1:10 000 населения (по разным данным).
Основой заболевания служит генетический дефект: мутация гена, ответственного за синтез белка соединительной ткани фибриллина. Ген расположен на 15-й хромосоме.
Наследуется заболевание по аутосомно-доминантному типу. Аутосомное означает, что оно не связано с полом, а доминантное – то, что мутация всегда проявляется. Степень проявления (распространенность нарушений и их выраженность) может варьироваться в различных пределах, что связано с генетическими особенностями.
Фибриллин придает соединительной ткани эластичность и растяжимость. Нарушение его строения приводит к утрате прочности и упругости соединительной ткани, и она перестает служить прочным каркасом. В первую очередь изменения касаются стенок сосудов, связочного аппарата. Даже небольшие физиологические нагрузки становятся запредельными для организма, соединительная ткань их не выдерживает.
Все случаи синдрома Марфана с точки зрения причины подразделяют на:
- семейные: составляют 75%, это передающаяся из поколения в поколение мутация гена;
- случайные (спорадические): 25%, это впервые возникшая мутация гена в роду, где ранее не было подобной патологии.
Клинических проявлений заболевания весьма много. Их рассматривают с точки зрения поражения отдельных органов и систем:
- опорно-двигательного аппарата;
- сердечно-сосудистой системы;
- органа зрения;
- нервной системы;
- бронхолегочной системы;
- кожи, мягких тканей;
- других органов.
Больные с синдромом Марфана обычно имеют высокий рост, худощавы из-за недоразвития подкожно-жировой клетчатки. Туловище при этом кажется коротким, а конечности – непропорционально длинными. Размах рук больше роста на 5% и более. Череп вытянутый (долихоцефалический), пальцы длинные паукообразные (арахнодактилия). Лицо вытянутое, узкое, небо высокое аркообразное (готическое), уши большие, нижняя челюсть выступает вперед (неправильный прикус), зубы растут неправильно, глаза глубоко посажены.
Деформация скелета заключается в наличии воронкообразной или килевидной грудной клетки, любых изменений в оси позвоночника (избыточный лордоз, кифозы, кифосколиозы, сколиозы), подвывихов позвонков (особенно в шейном отделе), спондилолистеза (смещение вышележащего позвонка по отношению к нижележащему). Характерно плоскостопие различных степеней выраженности, разболтанность суставов, избыточные движения в них (например, переразгибание), протрузия вертлужной впадины (углубление с глубоким погружением головки бедренной кости в области тазобедренного сустава). Обычно все это сопровождается снижением мышечного тонуса.
Изменения в этой системе часто являются наиболее тяжелыми, требующими коррекции в первую очередь, поскольку могут иметь угрожающий жизни характер.
Чаще всего синдром Марфана проявляется наличием пороков развития сердца и крупных сосудов (в частности, аорты). Дефекты строения сердца заключаются в нарушении перегородок сердца и его клапанов (пролапс митрального клапана, дефект межжелудочковой или межпредсердной перегородки, патологическое удлинение хорд). Нарушение строения стенки аорты приводит к развитию ее дилятации (расширению) и расслоению стенок (расслаивающая аневризма аорты). Из врожденных пороков крупных сосудов могут наблюдаться коарктация аорты, стеноз (сужение) легочной артерии.
Патологическое строение сердца и сосудов сопровождается развитием сердечной недостаточности, нарушением сердечного ритма (наджелудочковая и желудочковая тахикардии, фибрилляция предсердий и других), что весьма опасно для жизни больного. Присоединение инфекционных осложнений чревато развитием инфекционного эндокардита.

Симптомы поражения глаз очень специфичны для данного заболевания. Поражение опорно-двигательного аппарата, сердечно-сосудистой системы и глаз составляют типичную триаду симптомов при болезни Марфана.
Больные синдромом Марфана часто имеют голубые склеры. Зрение плохое ввиду развития близорукости высокой степени, длина глазного яблока увеличена. Зрачки могут быть не симметричными, то есть разными по размеру. Однако этим поражение органа зрения не ограничивается. Развиваются косоглазие, вывих или подвывих хрусталика, недоразвитие радужки и ресничных мышц, уплощение роговицы. Возможна отслойка сетчатки с развитием слепоты.
Поражение нервной системы в основном связано с патологией строения стенки сосудов. Это приводит к нарушению кровообращения, развитию ишемических или геморрагических инсультов (чаще – субарахноидальных кровоизлияний).
Возможны частые обмороки. Из аномалий возможны эктазия твердой мозговой оболочки, в частности пояснично-крестцовое менингоцеле (выпячивание оболочек мозга под кожу с образованием кармана через дефект в позвонках).
Возможны отклонения в психомоторном развитии, причем как в сторону задержки, так и в сторону превышения показателей. Многие люди с синдромом Марфана имеют высокие показатели интеллекта (выше, чем среднестатистический показатель IQ в популяции).
Еще одним симптомом при болезни Марфана могут служить гипофизарные расстройства с повышением содержания адреналина в крови, развитием акромегалии и несахарного диабета.
Поражение бронхов и легких заключается в развитии спонтанных пневмотораксов, дыхательной недостаточности, эмфиземы легких, апикальных булл (пузырьковидные образования в верхушках легких). Все эти изменения становятся результатом нарушения ангиоархитектоники легких из-за патологии соединительной ткани, повышенной растяжимости и пониженной эластичности легочной ткани.
Часто больные с синдромом Марфана имеют атрофические стрии на коже: волнистые полосы на коже разной ширины и цвета (от белого до красно-фиолетового). Малое количество подкожно жировой клетчатки становится причиной малого веса таких больных.
Больные с синдромом Марфана страдают рецидивирующими паховыми и бедренными грыжами.
Патология соединительной ткани становится причиной опущения почек, мочевого пузыря и матки, варикозного расширения вен, приводит к образованию длинного и со слабой перистальтикой кишечника (что сопровождается запорами).
Диагностика синдрома Марфана требует учета анамнестических данных (в том числе и наследственного анамнеза), данных осмотра больного, а также проведения разнообразных дополнительных методов исследования, позволяющих обнаружить изменения в сердце, сосудах, головном мозге, легких, глазах. Для этого могут понадобиться ЭКГ, ЭхоКГ (УЗИ сердца), рентгенография грудной клетки и тазобедренных суставов, КТ или МРТ головного и спинного мозга, сердца и сосудов, офтальмоскопия, аортография и другие методы. Перечень исследований зависит от наличия симптомов у конкретного больного.
Существуют диагностические критерии этого заболевания: большие и малые. Определенное сочетание больших и малых критериев и позволяет подтвердить диагноз. К большим критериям, например, относят подвывих хрусталика, эктазию твердой мозговой оболочки, килевидную или воронкообразную грудную клетку, плоскостопие, протрузию вертлужной впадины и другие. К малым критериям относят избыточную подвижность в суставах, готическое небо, деформацию черепа, спонтанный пневмоторакс и другие.
Окончательный диагноз выставляется после применения молекулярно-генетического метода и установления мутации гена, ответственного за синтез фибриллина.
Лечение синдрома Марфана всегда требует одновременного участия нескольких специалистов: кардиолога, кардиохирурга, офтальмолога, ортопеда-травматолога, терапевта.
Спектр лечебных процедур охватывает консервативные и оперативные способы лечения. Консервативные меры направлены на профилактику осложнений и поддержание нормального функционирования органов и систем, а оперативные предполагают коррекцию имеющихся анатомических изменений с целью предотвратить выраженное нарушение функций или даже угрозу для жизни больного.
Хирургические методы лечения:
- реконструктивные операции на аорте (при значительном, более 5 см, расширении восходящей части аорты и расслоении ее стенки);
- протезирование клапанов сердца;
- удаление измененного хрусталика с заменой его на искусственный;
- пластика позвоночника при выраженном сколиозе;
- эндопротезирование тазобедренных суставов;
- пластика грудной клетки (в последние годы отрицается ее целесообразность).
Больному необходим подбор очков или контактных линз, иногда возможна лазерная коррекция зрения.
Медикаментозное лечение имеет патогенетическую и симптоматическую направленность. Больному с метаболической целью назначают большие дозы витамина С (1-3 г в сутки), препараты с глюкозаминосульфатами и хондроитинсульфатами (Терафлекс, Структум, Хондроксид, Глюкозамин, Аминоартрин, Эльбона, Юниум), Карнитина хлорид 20% раствор, Янтарную кислоту по 100-200 мг 2 раза в день, препараты магния (например, Магне В6), поливитаминно-минеральные комплексы (с кальцием, магнием, цинком, медью). Прием этих веществ направлен на нормализацию обмена веществ, укрепление соединительной ткани.
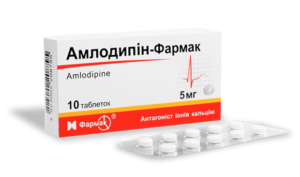 Для лечения сердечно-сосудистых нарушений часто используют β-адреноблокаторы (Пропранолол, Обзидан, Атенолол), блокаторы кальция (Нифедипин, Амлодипин, Лекоптин), ингибиторы ангиотензинпревращающего фермента (Лизиноприл, Периндоприл, Эналаприл), антиаритмические препараты (при нарушении ритма сердца). При развитии инфекционного эндокардита показаны антибиотики. После оперативных вмешательств может понадобиться антикоагулянтная терапия для снижения свертываемости крови (Фраксипарин, Клексан).
Для лечения сердечно-сосудистых нарушений часто используют β-адреноблокаторы (Пропранолол, Обзидан, Атенолол), блокаторы кальция (Нифедипин, Амлодипин, Лекоптин), ингибиторы ангиотензинпревращающего фермента (Лизиноприл, Периндоприл, Эналаприл), антиаритмические препараты (при нарушении ритма сердца). При развитии инфекционного эндокардита показаны антибиотики. После оперативных вмешательств может понадобиться антикоагулянтная терапия для снижения свертываемости крови (Фраксипарин, Клексан).
В целом подбор метода лечения и ассортимент применяемых лекарственных средств очень индивидуальны. Все зависит от спектра клинических симптомов и выраженности нарушений у конкретного больного.
Больным с синдромом Марфана показана лечебная физкультура, но в строго дозированном количестве, чтобы занятия приносили пользу сердечно-сосудистой и опорно-двигательной системам, а не вред. Нельзя заниматься контактными и игровыми видами спорта (баскетбол, футбол), рекомендовано плавание.
Заболевание неизлечимо, но постоянное динамическое наблюдение за больным, хирургическая коррекция сердечно-сосудистых, суставных и офтальмологических нарушений позволяют снизить риск для жизни, улучшить ее качество, позволить заниматься своей профессией. Некоторые больные не доживают до 40 лет, что связано с сердечно-сосудистыми осложнениями, в других случаях продолжительность жизни составляет 70 лет (особенно после проведения кардиохирургических операций).
Таким образом, синдром или болезнь Марфана – это наследственное заболевание, течение которого во многом зависит от тщательности медицинского наблюдения и своевременности оказания медицинской помощи. Обычно лечение требует привлечения многих специалистов, но такой комплексный подход позволяет избежать множества осложнений и повысить качество жизни больного.
источник
Болезнь Марфана относится к наследственной патологии соединительной ткани с аутосомно-доминантным типом наследования. При этом заболевании в патологический процесс вовлекаются многие жизненно важные органы: опорно-двигательный аппарат, сердечно-сосудистая, бронхолегочная и центральная нервная системы, глаза. Особое внимание обращает на себя диссоциация ростовых параметров: превышение длины тела (нередко на 4-6 стандартных отклонения) при резком отставании показателей массы.
В настоящем исследовании ставилась задача определить активность ряда гормонов сыворотки крови пробандов, оказывающих влияние на рост и развитие ребенка. Соматотропная функция гипофиза оценивалась на основании определения уровня СТГ в сыворотке крови больного радиоиммунологическим методом.
Принимая во внимание данные научных изысканий, согласно которым СТГ стимулирует рост тканей не непосредственно, а через образование вторичного агента — соматомедина, действующего на клеточном уровне, нами изучалась также соматомединовая активность сыворотки крови пробандов.
Представление о функциональном состоянии инсулярного аппарата поджелудочной железы было получено на основании определения активности иммунореактивного инсулина (ИРИ) сыворотки крови с помощью радио-иммуно-аналитического (РИА) набора (ВНР).
Учитывая результаты многочисленных научных изысканий о малой информативности показателей базального уровня пептидных гормонов, изучение активности СТГ и инсулина сыворотки крови больных проводилось под контролем стандартного глюкозотолерантного теста (СГТТ). Нагрузка глюкозой у лиц с болезнью Марфана проводилась из расчета общепринятой дозировки и составляла 1,75 г/кг массы тела пробанда. Исследуемая сыворотка включала четыре пробы: тощаковую, затем через 1, 2 и 3 часа после приема глюкозы per os. Показатели уровней глюкозы в сыворотке крови измерялись ортотолуидиновым методом.
По клинической картине выделялись две группы пробандов: с полным и неполным симптомокомплексом болезни Марфана.
Анализ показателей активности СТГ в четырех пробах сыворотки крови пробандов позволил выделить две группы больных, отличающихся между собой по характеру ответной реакции гормона на нагрузку глюкозой. Первая группа включала 9 детей; ей было свойственно парадоксальное увеличение активности соматотропина на СГТТ. Базальный уровень СТГ в сыворотке крови у большинства пациентов первой группы был низким и не превышал 3,8 нг/мл (1 больной) при норме 3,81±0,2 нг/мл. Дальнейший анализ показателей активности СТГ в процессе СГТТ констатировал их повышенные значения у всех 9 детей. Полученные результаты были достаточно весомы и у 3 больных соответствовали значениям 20, 25,8 и 33,3 нг/мл.
Вторую, большую группу составили 16 пробандов. Активность гормона на нагрузку глюкозой у больных второй группы существенно подавлялась, что соответствовало норме. Базальный уровень соматотропина был повышен у 6 пациентов и у 3 из них составлял 15,8, 20 и 20 нг/мл.
Таким образом, изучение показателей активности СТГ сыворотки крови у лиц с болезнью Марфана выявило их превышение у 15 пациентов, причем, наиболее высокие цифры зафиксированы у детей первой группы, патологически реагировавшей на СГТТ. Необходимо подчеркнуть, что все пробанды с парадоксальной реакцией на СГТТ имели полный симптомокомплекс болезни Марфана.
Изучение соматомединовой активности сыворотки крови проводилось у 5 больных. Отчетливое повышение активности соматомединов до 1 ед/мл выявлено лишь у 1 пробанда (Иван Т., 19 лет), у 1 пациента 16 лет активность гормона соответствовала верхним значениям нормы и составляла 0,88 ед/мл. У трех детей соматомединовая активность сыворотки крови была низкой (0,1 ед/мл, 0,35 ед/мл, 0,39 ед/мл).
Определение активности иммунореактивного инсулина (ИРИ) в сыворотке крови больных при нагрузке глюкозой проводилось у 16 пробандов. Обращал на себя внимание тот факт, что повышение уровня ИРИ под влиянием СГТТ происходило почти у всех больных, однако, степень выраженности этих изменений была большей у детей с полным симптомокомплексом болезни Марфана и достигала максимальных значений 98,5 мкмЕ/мл (Дина С. 16 лет) (при норме 10-20 мкмЕ/мл).
При анализе изменений толерантности к глюкозе у пробандов с болезнью Марфана, наряду с нормальной реакцией (12) выявлена патология, преимущественно двух типов: предиабетические (4) и гипореактивные или плоские (9) сахарные кривые. При этом оба вида нарушений углеводного обмена констатированы у пациентов с полным симптомокомплексом заболевания.
Проведенное нами изучение состояния щитовидной железы, принимающей участие в процессах регуляции роста и развития, не выявило отклонений. Анализ показал физиологическое повышение уровней Т3 и Т4 у этих больных и полное отсутствие ответного выброса гипофизом СТГ на рилизинг-фактор ТТГ. Полученные нами данные полностью соответствовали исследованиям Hainer, не обнаружившим увеличения секреции СТГ в ответ на стимуляцию рилизинг-фактором ТТГ у здоровых лиц разного возраста.
Таким образом, в результате проведенных исследований изменений тиреотропной функции гипофиза и щитовидной железы обнаружено не было. Основные нарушения касались соматотропного гормона, ИРИ и толерантности тканей больных к глюкозе.
На основании статистических данных становится очевидным, что повышение уровня СТГ констатировано у 15 пробандов с болезнью Марфана, соматомедина — у 1, ИРИ — у 10 больных. Изменения типов сахарных кривых отмечены у 13 пациентов. Снижение базальной активности СТГ сыворотки крови зафиксировано у 18 больных, соматомедина — у 3, ИРИ — у 13. Нормальные значения соматотропина, соматомедина и ИРИ выявлены только у 1 пробанда.
Сравнительный анализ полученных результатов, отражающих состояние органов внутренней секреции с тяжестью клинической симптоматики основного заболевания и другими показателями метаболизма (соединительной ткани, репарации повреждений ДНК, основного обмена и др.) выявил четкую зависимость между изученными параметрами. Так, дети с полным симптомокомплексом синдрома Марфана имели более выраженную клинику болезни, проявляющуюся в ранней и тяжелой манифестации заболевания, быстром его прогрессировании и развитии необратимых инвалидизирующих расстройств. Роман Л. в 3 года опережал своих сверстников в длине тела на 3,5 стандартных отклонения при низких показателях массы. Фенотип ребенка обращал на себя внимание тяжелыми изменениями опорно-двигательного аппарата (врожденная воронкообразная деформация грудной клетки третьей степени, кифосколиоз, плоскостопие, арахнодактилия, мышечная гипотония), глаз (миопия, крайне высокой степени), расстройством сердечно-сосудистой деятельности с признаками декомпенсации. Показатели почечной экскреции оксипролина у больного превышали возрастную норму в 4 раза. Результаты СГТТ были необычны и характеризовались отсутствием подавления активности СТГ, нулевыми значениями инсулина и предиабетической сахарной кривой. У Гали Ш. в 6 лет, наряду с типичной клиникой поражения скелета и глаз имелись четкие признаки расширения корня аорты и пролапса митрального клапана. Данные почечной экскреции оксипролина у больной в 2,5 раза превышали средние значения нормы, а показатели основного обмена достигали особенно высоких цифр и составляли +114%. Сахарная кривая ребенка свидетельствовала о предиабетических изменениях углеводного обмена. Ответная реакция СТГ на СГТТ носила патологический характер.
Наташа К. в 12 лет имела длину тела 178 см, миопию -15 Д (вследствие подвывиха хрусталиков), пролапс митрального клапана и расширение корня аорты. Показатели почечной экскреции оксипролина в три раза превышали норму. Сахарная кривая была плоской. Активность СТГ достигала 33,3 нг/мл, а величина основного обмена превышала норму на 73%. Заболевание у Васи П. (16 лет) характеризовалось ранней манифестацией, тяжелым поражением многих органов и систем: полной слепотой, декомпенсацией сердечно-сосудистой системы, требующей постоянного применения медикаментозных средств, нарушениями эмоционально-волевой сферы. Наряду с парадоксальной реакцией СТГ на СГТТ, его репарационные показатели повреждений ДНК были наиболее низкими и составляли только 51% против 97% у здорового ребенка.
Больные с неполным симптомокомплексом характеризовались, как правило, более легким течением- основного заболевания. Так, например, у Ивана Т., Вовы и Александра Н., Славы П. и др. не был диагностирован подвывих хрусталиков. Эндокринная функция у этих больных страдала в меньшей степени: отмечалась физиологическое подавление активности СТГ на СГТТ, нормальные колебания уровня ИРИ сыворотки крови и нормореактивные типы сахарных кривых, их репарационные возможности повреждений ДНК показывали наиболее высокие результаты (89% У Ивана Т.), а показатели основного обмена были относительно низкими ( + 8% у Славы П.).
Таким образом, исследования, проведенные нами, четко констатировали вовлечение в патологический процесс органов эндокринной системы при болезни Марфана и, в первую очередь, соматотропной функции гипофиза и инсулярного аппарата поджелудочной железы. Становится очевидным, что эндокриные расстройства не только усугубляют тяжесть основного заболевания, но в ряде случаев являются, вероятно, одним из основных патогенетических механизмов формирования кардинальных симптомов болезни. Выраженные изменения гормональных функций, обнаруженные нами у детей младшего возраста с полным тяжелым симптомокомплексом болезни Марфана, свидетельствуют в пользу первичного поражения органов эндокринной системы при этом заболевании. Так, например, известно, что некоторые крупные молекулы сывороточных белков вызывают пролиферацию мышечных клеток среднего слоя аорты. Аналогичные изменения больших сосудов установлены под воздействием СТГ, при этом предполагается непосредственное влияние гормона на сосудистую стенку. Авторы сообщают также о возможном участии соматотропина в развитии диабетических микроангиопатий. Не исключено, что в возникновении тяжелых сердечно-сосудистых расстройств при болезни Марфана (аневризма аорты и других сосудов, пролапс митрального клапана) действию СТГ отводится главная роль.
Эффект соматотропина на рост кости определяется по изучению почечной экскреции оксипролина. Результаты, отражающие обмен коллагеновых волокон, полученные нами ранее, четко соответствуют показателям активности СТГ: высокий уровень гормона регистрируется, примерно, у 3/5 обследованных больных (15 из 25). Однако, клиническая симптоматика болезни Марфана, наряду с изменениями углеводного и основного обменов, свидетельствуют о возможном повышении уровня соматотропина у большинства пациентов с этой тяжелой патологией соединительной ткани. Merimel с соавт. при обследовании больных с изолированной недостаточностью СТГ выделяют группу детей, реагировавших гиперсекрецией инсулина в ответ на глюкозу. Исследователи выдвигают гипотезу, согласно которой эти больные секретируют СТГ с измененной и радиоиммунологически не определяемой молекулой. В связи с этим логично предположение об идентичном нарушении в строении гормона роста у некоторых пробандов с болезнью Марфана. Выявленная нами гиперсекреция инсулина на СГТТ при низких показателях гормона роста служит косвенным подтверждением такой гипотезы. Нельзя также исключить вероятность увеличенной биологической активности эндогенного СТГ у пациентов с болезнью Марфана. Предположение о высокой биологической активности экзогенного СТГ высказывается Illig получившей выраженный ростовой эффект при лечении больных с высоким титром антисоматотропных антител препаратами СТГ. Следует также принять во внимание мнение Лашас Л. и Лашене Д. о значительном увеличении гормональных рецепторов в клетках больных при патологических состояниях.
Трудность интерпретации нарушений углеводного обмена при болезни Марфана объясняется неоднородностью этих изменений, обусловленных, по всей вероятности, различными патогенетическими механизмами. Так, низкая активность инсулина у некоторых детей, по-видимому, свидетельствует об истощении инсулярного аппарата поджелудочной железы, возникающей вследствие повышенной секреции СТГ при болезни Марфана. Аналогичные изменения обмена происходят у больных акромегалией. Преимущественное включение в энергетический обмен свободных жирных кислот, обусловленное у больных акромегалией повышенной продукцией СТГ, сопровождается уменьшением утилизации глюкозы и длительным усилением секреции инсулина, что в конечном итоге приводит к резкому уменьшению резервных возможностей железы. Казалось, что низкие значения активности инсулина плазмы, выявленные у детей с болезнью Марфана, должны были бы послужить причиной развития у них сахарного диабета, как это нередко бывает при акромегалии. Однако, ни в одном из 83 наблюдавшихся нами (в течение 12 лет) случаев заболевания не было найдено симптомов сахарного диабета. Тщательный анализ литературы позволил обнаружить лишь единичное сообщение о сочетании болезни Марфана и сахарного диабета. Принимая во внимание данные научных изысканий об общем механизме действия инсулина и соматомединов, не исключено, что ключ к частичному решению вопроса об отсутствии сахарного диабета у лиц с болезнью Марфана может быть найден при изучении соматомединовой активности сыворотки крови у этих больных. Небольшое количество исследований, проведенных нами в этом направлении, неоднородный возрастной состав больных и различная степень тяжести заболевания у пробандов не позволяют сделать пока достоверных выводов из этой работы. Нельзя также, по-видимому, не учитывать вероятность повышенного усвоения глюкозы тканями (плоские сахарные кривые) и возможность компенсаторного увеличения секреции соматостатина у детей с болезнью Марфана. Введение соматостатина больным акромегалией приводит к быстрому снижению уровня СТГ (на 50- 75%) и инсулина, что дает основание авторам рекомендовать этот гормон для лечения подобных заболеваний.
Таким образом, становится очевидной необходимость дальнейшего изучения показателей активности СТГ и инсулина плазмы совместно с рядом метаболических параметров, контролируемых данными гормонами. Получение этих результатов будет способствовать более углубленному изучению патогенетических механизмов болезни Марфана и, следовательно, назначению адекватной терапии. В связи с этим представляют интерес исследования Casanueva и соавт., доказавших участие НЭЖК в регуляции секреции СТГ вплоть до полной ее блокады при концентрации НЭЖК выше 3 мэкв/л. Работа испанских авторов подтверждает патогенетическую обоснованность лечения детей с болезнью Марфана энпитами с высоким содержанием жира. Положительный эффект, достигнутый С. М. Барашневой при диетотерапии таких детей высоко жировыми энпитами, еще раз подчеркивает необходимость включения данного препарата в комплекс терапевтических мероприятий, рекомендуемых при болезни Марфана.
Применение соматостатина — гормона-ингибитора, на который возлагаются большие надежды в лечении целого ряда заболеваний (язвенная болезнь желудка, акромегалия и др.) также, вероятно, сможет сыграть немалую роль в эффективной терапии болезни Марфана.
источник