Гемоглобинопатии преимущественно поражают население тропических и субтропических областей (Экваториальная Африка, Аравийский полуостров, Южная Индия, Южный Китай, Средиземноморье, Азербайджан, Грузия). Наиболее распространены и отличаются тяжестью проявлений серповидноклеточная анемия и большая талассемия, или анемия Кули. Большинство гемоглобинопатий клинически не проявляются; при некоторых — могут наблюдаться: анемия, эритроцитоз или цианоз (например, при метгемоглобинопатиях).
а) Нарушения первичной структуры глобиновых цепей гемоглобина вследствие генной мутации. Описано около 500 аномальных гемоглобинов.
Тип наследования аутосомно-рецессивный, дефект бета-глобиновых цепей, в которых в 6 положении гидрофильный глютамин заменен гидрофобным валином, образуется HbS. Восстановленная форма HbS мало растворима. При гипоксемии и снижении скорости кровотока HbSполимеризуется в длинные нерастворимые нити, растягивающие эритроциты в форме серпа. Если содержание HbS больше 45% (гомозиготное состояние), образуются эритроциты необратимо серповидной формы, склонные к агрегации, что повышает вязкость крови, вызывает закупорку сосудов (вазоокклюзию), нарушение микроциркуляции и боль.
В этом случае точечная мутация обусловливает нарушение структуры гемоглобина, гемолиз, анемию, а также закупорку сосудов и нарушение кровообращения.
Больные СКА имеют типичный вид: удлиненный нижний сегмент тела, выступающий лоб, «башенный» череп, гепато-спленомегалия. Самый характерный криз для этого заболевания — вазоокклюзионный, проявляющийся резкой болью.Окклюзия сосудов может развиваться в разных органах, поэтому клинические симптомы чрезвычайно разнообразны. В мазках крови можно обнаружить серповидные клетки.
Примерно одна треть обитателей тропических и субтропических регионов Африки (т. наз. «малярийного пояса») являются носителями признака серповидноклеточности. Они не страдают от гемолитической анемии, в то же время более устойчивы по отношению к малярии.
Талассемии — замедление или отсутствие синтеза одной из цепей глобина:
При талассемиях мутации располагаются не в структурных генах, а в генах-регуляторах, поэтому структурных нарушений нет, а результатом мутаций служит замедление или отсутствие синтеза одной из глобиновых цепей и замена ее синтезом другой цепи. Талассемия встречается в странах Средиземноморья, в Китае, Индии, в Европе, у жителей Закавказья и Средней Азии. Самая высокая заболеваемость – на Мальдивах, где носители признака составляют 18%. Гетерозиготная бета-талассемия наблюдается у 7— 10% населения в низменных районах Азербайджана.
Альфа-талассемия – полное или частичное прекращение синтеза α-цепей. Компенсаторно синтезируются: а) в пренатальный период γ-цепи — образуется тетрамер γ (Hb Барт); б) в постнатальный – тетрамер ß (HbH).
Синтез α-цепей кодируют 4 гена, поэтому степень нарушения их синтеза меньше, чем при ß-талассемии; выраженный дисбаланс развивается только тогда, когда поражены все 4 гена. Агрегаты из ß-цепей более растворимы, чем агрегаты из α -цепей, поэтому гемолиз при α-талассемии выражен слабее, чем при ß-талассемии, а эритропоэз более эффективен.
Бета-талассемияобусловлена снижением скорости синтеза ß-цепей гемоглобина (ß + -талассемия) или отсутствием их синтеза (ß 0 -талассемия). Неповрежденные α-цепи избыточно накапливаются в клетках, что ведет к повреждению мембраны и разрушению эритроидных клеток в костном мозге (неэффективный эритропоэз)и эритроцитов в крови. Деструкция эритроидных клеток способствует гиперплазии костного мозга, что отражается на структуре скелета, ведет к повышенному всасыванию железа и перегрузке организма железом.
Тяжелая гомозиготная форма ß-талассемии — болезнь Кули, или большая талассемия. Кроме того, выделяют промежуточную, малую и минимальную талассемию.
 Для тяжелых форм талассемий характерна:
Для тяжелых форм талассемий характерна:
желтушность, хронические язвы на нижних конечностях, башенный череп,
уплощенная переносица. Скулы выступают, глазные щели сужены, нарушены прикус и расположение зубов.
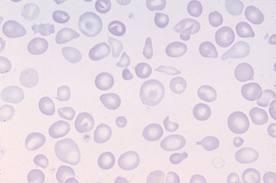
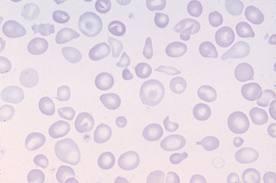
 Картина крови: мишеневидные эритроциты, анизоцитоз, пойкилоцитоз
Картина крови: мишеневидные эритроциты, анизоцитоз, пойкилоцитоз
Сканирующая электронограмма. Стрелками показаны два кодоцита («хвостатые клетки») – это другое название мишеневидных клеток.
источник
 Анемия Под определение «гемолитическая анемия» подпадает любая анемия, характеризующаяся преобладанием гемолиза (процессом распада эритроцитов) над эритропоэзом (процессом образования эритроцитов). Гемолиз может быть иммунный и неиммунный. Гемолитическая анемия бывает наследственные и приобретенные.
Анемия Под определение «гемолитическая анемия» подпадает любая анемия, характеризующаяся преобладанием гемолиза (процессом распада эритроцитов) над эритропоэзом (процессом образования эритроцитов). Гемолиз может быть иммунный и неиммунный. Гемолитическая анемия бывает наследственные и приобретенные.
- Наследственная форма гемолитической анемии, обусловленная нарушением мембраны эритроцитов
- Наследственная форма гемолитической анемии, обусловленная нарушением активности ферментов эритроцитов
- Наследственная форма гемолитической анемии, обусловленная нарушением синтеза или структуры гемоглобина
- Анемия, обусловленная влиянием антител
- Анемия, обусловленная изменением структуры мембраны, вызванной соматической мутацией
- Анемия, обусловленная механическим повреждением оболочки эритроцитов
- Анемия, вызванная химическим повреждением эритроцитов
- Анемия, вызванная дефицитом витаминов (фолиевой кислоты и цианокобаламина)
- Анемия, вызванная разрушением эритроцитов паразитами
Болезнь Минковского-Шоффара (наследственный микросфероцитоз) – группа наследственных гемолитических анемий, характеризующихся образованием микросфероцитов (шаровидных эритроцитов) и обусловленных дефектом протеинов цитоскелета эритроцитов. При этом эритроциты теряют часть мембраны, уменьшается соотношение площади поверхности к объему, в результате чего эритроцит превращается в микросфероцит. Как правило, патология наследуется по аутосомно-доминантному признаку. Распространенность наследственного микросфероцитоза составляет примерно 1 случай на 1000-4500 человек.
При наследственном микросфероцитозе генетические нарушения влияют на протеины цитоскелета, преимущественно на те, которые объединяют цитоскелет с мембраной эритроцита. У большинства больных отмечается значительный дефицит спектрина, и только в некоторых случаях этот дефицит обусловлен генетическими дефектами самого спектрина.
Главные признаки наследственного микросфероцитоза – анемия, желтуха, спленомегалия (увеличенная селезенка). Анемия возникает из-за внутриклеточного распада эритроцитов. Желтуха развивается посредством непрямой гипербилирубинемии, может быть непостоянной и, как правило, слабо выражена у детей раннего возраста. Повышенное содержание билирубина в желчи часто является причиной образования пигментных желчных камней (даже у детей). Увеличение селезенки (спленомегалия) отмечается практически во всех случаях. При системных инфекционных патологиях интенсивность гемолиза может увеличиваться, в результате чего развивается спленомегалия.
Тяжелые формы наследственного микросфероцитоза характеризуются деформацией скелета: изменение расположения зубов, акрокефалия (башенный череп), высокое верхнее небо, микрофтальмия (уменьшение глазного яблока). В некоторых случаях отмечаются укороченные мизинцы. Могут образовываться трофические язвы на ногах.
Наследственный микросфероцитоз сопровождается апластическими кризами, которые провоцируются инфекцией (особенно парвовирусной).
Микросфероцитоз – характерное изменение формы эритроцитов при этой патологии. При анализе мазка крови в биологическом материале наблюдаются микросфероциты в виде мелких клеток без центрального просветления (см рисунок 1). Отметим, что обнаружение микросфероцитов в мазках не всегда является признаком наследственного сфероцитоза.
Рисунок 1. Наследственный микросфероцитоз. Микросфероциты в мазке периферической крови (окр. по Романовскому-Гимзе, ув. ×100)
Такой признак обнаруживается при аутоиммунной гемолитической анемии с неполными тепловыми агглютинами, при наследственных дизэритропоэтической анемии. Средний объем эритроцитов, как правило, остается в норме или незначительно снижен. Показатель среднего содержания гемоглобина в эритроцитах в норме или незначительно повышен. Средняя концентрация гемоглобина в эритроцитах повышена почти у 50% пациентов.
Количественным показателем сферичности эритроцитов является осмотическая устойчивость (она снижена). Уровень ретикулоцитов в крови при гемолитическом кризе может значительно повышаться. Миелограмма показывает резкое раздражение красного ростка. Дифференциальный диагноз проводят с аутоиммунной гемолитической анемией, для которой характерна положительная проба Кумбса, отсутствие этой патологии среди родственников пациента и отсутствие данных о начале заболевания в детском возрасте.
Основной метод лечения анемии при наследственном микросфероцитозе – спленэктомия, с помощью которой устраняется анемия; при этом нельзя устранить морфологический дефект эритроцитов.
Наследственная гемолитическая анемия, обусловленная дефицитом глюкозо-6-фосфат дегидрогеназы эритроцитов – наиболее распространенная ферментопатия эритроцитов из группы ферментопатий пентозофосфатного пути метаболизма глюкозы. Глюкозо-6-фосфатдегидрогеназа эритроцитов – олигомер (в зависимости от условий может быть димер или тетрамер), который состоит из субъединиц с молекулярной массой 56 000 D. По данным ВОЗ (Всемирной организации здравоохранения) во всем мире количество людей, страдающих этой патологией, составляет более 200 млн. Наиболее широкое распространение этого заболевания характерно для Средиземноморского региона (Сицилия, Греция, Сардиния), негроидной расы, жителей Ближнего и Дальнего востока.
Клиническая картина при наследственной форме гемолитической анемии полиморфна: степень тяжести патологии может колебаться от гемолитической анемии, возникающей спонтанно после рождения, до гемолитических кризов. Гемолитический криз, который может провоцироваться метаболическим ацидозом или гипогликемией, развивается за несколько часов. В тяжелых случаях у больного развивается гемоглобинурия и шок. Также наблюдаются желтуха, моча приобретает бурый или черный цвет, одышка, диарея, рвота, снижение артериального давления, развивается тяжелая анемия, увеличиваются печень (гепатомегалия) и селезенка (спленомегалия).
Тяжелый гемолитический криз может спровоцировать развитие ДВС-синдрома (диссеминированного внутрисосудистого свертывания крови). Некоторые пациенты не переносят конские бобы (Viciafaba), после употребления которых происходит молниеносное развитие гемолитического криза (это явление также известно, как фовизм или примахиновая анемия).
Дефицит глюкозо-6-фосфат дегидрогеназы эритроцитов необходимо подозревать во всех случаях острого гемолиза, особенно у лиц негроидной расы и жителей средиземноморского региона. Диагноз подтверждается путем проведения лабораторных анализов. Острый гемолиз характеризуется быстрым снижением гематокрита с одновременным повышением уровня гемоглобина и непрямого гемоглобина, а также снижением уровня гаптоглобина. Анализ мазка крови показывает наличие фрагментов эритроцитов. Основой диагностики считается качественное (при необходимости – количественное) определение активности глюкозо-6-фосфат дегидрогеназы эритроцитов. У пациентов с вариантом «А-» явление аномального гемолиза проходит, как правило, самостоятельно – такие больные не нуждаются в специальном лечении. В случае развития тяжелого гемолитического криза необходимо проводить форсированный диурез, профилактику ДВС-синдрома, плазмаферез (с целью удаления продуктов гемолиза).
В случае возникновения качественной гемоглобинопатии происходит изменение аминокислотной последовательности цепей глобина. Талассемия (количественная гемоглобинопатия) характеризуется снижением образования цепей глобина без изменения их цепей. Нужно отметить, что разница между качественной и количественной гемоглобинопатиями не абсолютна.
Талассемия (анемия Кули) – группа патологий, обусловленных генетическим нарушением синтеза одной из цепей глобина. В норме процесс синтеза глобиновых цепей сбалансирован, поэтому свободных цепей глобина нет. В случае нарушения синтеза одной из цепей глобина баланс нарушается, образуются лишние цепи, которые агрегируют и откладываются в эритрокариоцитах. Среди жителей Средиземноморья наиболее распространена β-талассемия.
«Большая талассемия» (болезнь Кули, β-талассемия) – наследственная гемолитическая анемия, впервые описанная американскими педиатрами-гематологами Томасом Бентоном Кули (Thomas Benton Cooley) и Ли (P. Lee) в статье «Серия случаев спленомегалии у детей с анемией и необычными изменениями костей» («A Series of Cases of Splenomegaly in Children, with Anemia and Peculiar Bone Changes»), где были приведены случаи у выходцев из стран Средиземноморья. Для анемии Кули характерна тяжелая степень течения с самого детства, задержка роста и изменения костей в результате увеличения объема костного мозга, возникающие в случае отсутствия соответствующего лечения). Также при этой патологии у больного наблюдаются гепатомегалия, спленомегалия, гиперспленизм, деформации черепа (монголоидное лицо, башенный череп); желтуха, бледность и отложение меланина придают коже особый медный оттенок. Кроме этого, наблюдается перегрузка железом сердца, легких, печени, поджелудочной железы и других органов эндокринной системы, переломы костей, сдавления периферических нервов, разного рода инфекционные осложнения.
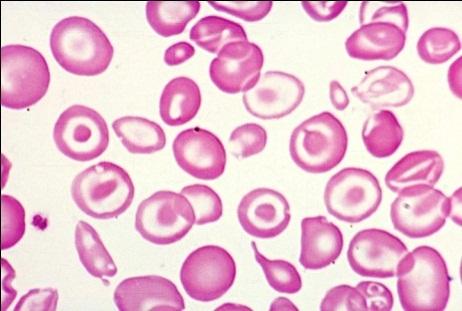
Результаты лабораторных исследований периферической крови показывают гипохромную анемию, ретикулоцитоз, мишеневидные эритроциты (см рис 2-4).
Рисунок 02. Анемия Кули (большая талассемия). Периферическая кровь. Микроцитоз, выраженная гипохромия, мишеневидные нормобласты и эритроциты (окр. по Романовскому-Гимзе, ув. ×100)
Рисунок 03. Анемия Кули (большая талассемия). Периферическая кровь (окр. по Романовскому-Гимзе, ув. ×50)
Рисунок 04. Анемия Кули (большая талассемия). Периферическая кровь. Множественные мишеневидные эритроциты (окр. по Романовскому-Гимзе, ув. ×100)
Миелограмма демонстрирует раздражение «красного ростка» и повышение количества сидеробластов. Также наблюдается повышение осмотической резистентности эритроцитов и количества билирубина за счет непрямой фракции. В крови повышается содержание железа и ферритина, развивается гемосидероз (чрезмерное отложение гемосидерина в тканях) внутренних органов. При гомозиготной β-талассемии необходимо проводить пренатальную диагностику – забор клеток плода из амниотической жидкости на предмет выявления мутации генов, отвечающих за кодирование β-цепи глобина, с применением метода полимеразной цепной реакции.
Без соответствующего лечения больные анемией Кули умирают в детском возрасте. Продлить жизнь, предупредить деформации костей и задержку роста можно путем регулярных трансфузий эритроцитарной массы (лучше переливать отмытые или размороженные эритроциты) при условии поддержания достаточно высокого уровня гемоглобина. В случае значительной спленомегалии и явлениях гиперспленизма больному показана спленэктомия (удаление селезенки). С целью предотвращения развития гемосидероза пациентам периодически назначают Деферазирокс (Эксиджад) или Дефероксамин (Десферал). Излечение возможно при аллогенной трансплантации костного мозга.
Серповидноклеточная анемия обусловлена носительством гемоглобина, который меняет свою структуру в условиях гипоксии. Самой распространенной аномалией структуры гемоглобина является гемоглобинопатия Sα2β26 глу+вал. При гомозиготном носительстве можно говорить о серповидноклеточной анемии; при гетерозиготном носительстве – серповидноклеточная аномалия. Патология наследуется по аутосомно-доминантному признаку. При серповидноклеточной анемии наблюдается мутация, в результате которой в цепи глобина глутаминовая кислота заменяется валином. В результате растворимость гемоглобина S при отдаче кислорода снижается, что приводит к образованию геля.
Серповидноклеточная анемия наиболее распространена среди населения Центральной Африки, Турции, Индии, Кубы. У больных диагностируется анемия, тромботические осложнения, поражения костей и суставов (отмечаются некрозы плечевой и бедренной костей). Кроме этого, тромбозы осложняются инфарктами (сердца, легких, почек, селезенки, головного мозга), приступами сильной боли в области живота. У детей отмечаются нарушения физического (отставание в росте) и полового развития, ночное недержание мочи, нарушение зрения (тромбозы сосудов сетчатки). Также могут развиваться гемолитический, апластический и секвестрационные кризы, при этом в селезенке происходит резкое накопление эритроцитов, что вызывает гиповолемический шок и резкое снижение уровня гемоглобина.
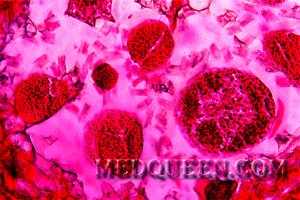
Для анализов крови при апластической анемии характерны низкий уровень гемоглобина, наличие серповидных эритроцитов (рисунок 5), базофильная пунктация эритроцитов, их мишеневидность, повышение уровня ретикулоцитов и непрямого билирубина. Миелограмма демонстрирует раздражение «красного ростка».
Рисунок 5. Серповидноклеточная анемия. Периферическая кровь. Серповидные и мишеневидные эритроциты. выраженная гипохромия эритроцитов (окр. по Романовскому-Гимзе, ув. ×100)
В качестве лечения применяют адекватную инфузионную терапию, переливания эритроцитарной массы, оксигенотерапии.
К приобретенным гемолитическим анемиям относится группа заболеваний разного патогенеза, которые объединяет внутрисосудистый гемолиз (гемолиз эритроцитов в периферической крови). В зависимости от механизма эритролиза приобретенная гемолитическая анемия может носить иммунный и неиммунный характер. Но, несмотря на разные патогенетические механизмы, клинические признаки этих анемий часто совпадают.
Гемолитическая анемия у пациентов с протезированными клапанами сердца и сосудами развивается примерно в 10% случаев при протезированном аортальном клапане. При использовании стеллитовых запирательных элементов частота гемолиза незначительно увеличивается (по сравнению с селиконовыми). Также некоторое увеличение частоты гемолиза отмечается при наличии околоклапанной регургитации и при малом диаметре клапана. Биопротезы (свиные клапаны) в редких случаях являются причиной механического гемолиза. Гораздо реже причиной гемолиза может быть также протезированный митральный клапан, так как трансклапанный градиент давления в этом случае ниже.
Гемолиз протезированными клапанами происходит в результате одновременного действия сразу нескольких факторов:
- Значительная сила сдвига, которая при турбулентном токе крови действует на мембрану эритроцитов, особенно когда под высоким давлением кровь проходит через маленькое отверстие (например, при околоклапанной регургитации)
- Отложения фибрина на участках неплотного прилегания кольца клапана к тканям сердца
- Прямое механическое повреждение эритроцитов при закрытии запирательного элемента
Значительное разрушение эритроцитов может наблюдаться после закрытия дефекта межпредсердной перегородки типа ostium primum заплатой из синтетического материала. Умеренное сокращение жизни эритроцитов с легкой анемией или без нее может наблюдаться при значительном обызвествлении аортального клапана. Механический гемолиз обнаруживается также у пациентов, перенесших аортокоронарное и аортобедренное шунтирование.
Тяжелые случаи механического гемолиза сопровождаются тяжелой анемией, ретикулоцитозом, обнаруживаются фрагментированные эритроциты (шизоциты), гемоглобинемия и гемоглобинурия, повышается активность лактатдегидрогеназы, снижается уровень гаптоглобина. Выведение железа из организма с мочой в виде гемосидерина или гемоглобина может вызвать дефицит железа в организме. В случае развития дефицита железа пациенту назначается пероральный прием препаратов железа. Терапия препаратами железа способствует повышению уровня гемоглобина и способствует снижению сердечного выброса и снижению интенсивности гемолиза. Отметим, что ограничение физической активности также способствуют снижению интенсивности распада эритроцитов. Если предпринимаемые меры не приводят к желаемому результату, нужно полностью устранить околоклапанную регургитацию или заменить протез.
источник
Северо-Западный государственный медицинский университет имени И.И.Мечникова
Министерства здравоохранения Российской Федерации
(ГБОУ ВПО СЗГМУ им. И.И.Мечникова Минздрав России)
Кафедра факультетской терапии
Т.В. Ермолова
ГЕМОЛИТИЧЕСКИЕ АНЕМИИ
(УЧЕБНО-МЕТОДИЧЕСКОЕ ПОСОБИЕ ДЛЯ СТУДЕНТОВ 4 КУРСА)
Обсуждено на заседании кафедры
№протокола7 дата29 января 2015г.
Тема: Гемолитические анемии. Место проведения: больничная палата и учебный класс.
Продолжительность темы: 4 часа.
Изучить особенности клинической картины, течения, диагностики, лечения гемолитических анемий, а также определить прогноз больного.
· Определение гемолитических анемий
· Классификацию гемолитических анемий
· Этиологию гемолитических анемий
· Современные представления о патогенезе гемолитических анемий
· Основные клинические проявления гемолитических анемий
· Методы верификации диагноза гемолитических анемий
· Современные методы лечения больных с гемолитическими анемиями
· Определение прогноза больного
· методически правильно собрать и интерпретировать жалобы и анамнез
· провести физикальное исследование больного
· сформулировать предварительный диагноз и обосновать его на основании клинических данных
· составить план обследования больного с учетом необходимости: а) верификации диагноза б) определения состояния органов и систем в) дифференциального диагноза основных клинических проявлений заболевания
· правильно оценить результаты исследований
· сформулировать клинический диагноз у больного с гемолитической анемией и обосновать его
· составить схему лечения больного
· оценить прогностическую значимость заболевания
Гемолитические анемии составляют около 5% среди всех заболеваний крови, а среди анемических состояний — 11,5 %. В структуре гемолитических анемий преобладают наследственные формы заболеваний.
Вопросы для самоподготовки.
· Определение понятия гемолитической анемии
· Классификация гемолитических анемий
· Основные жалобы и клинические проявления при гемолитической анемии
· Патогенез гемолитических анемий
· Принципы лечения гемолитических анемий
· Прогноз больного при гемолитической анемии
Задания для самоподготовки
· Понятие о внутриклеточном и сосудистом гемолизе
· Тактика лечения больных с гемолитическими анемиями
1.http://www.who.int/classifications/apps/icd/icd10online/?kd50.htm (классификация МКБ-10)
2.Мамаев Н.Н., Рябов С.И.Гематология: Руководство для врачей.-Москва: СпецЛит.,2008.-560 с.
3.Воробьев А.И.Руководство по гематологии.-Москва:Ньюдиамед, 2005.-416 с.
4.GriffinP.Rodgers, Neal S.Young.The Bethesda Handbook of Clinical Hematology, 2013.-512 p.
Определение.
Гемолитические анемии — группа достаточно редко встречающихся заболеваний, общим признаком которых является усиленное разрушение эритроцитов, обусловливающее, с одной стороны, анемию и повышенное образование продуктов распада эритроцитов, с другой стороны — реактивно усиленный эритропоэз.
Гемолитические анемии представляют собой клинико-гематологический синдром, характеризующийся укорочением продолжительности жизни эритроцитов в связи с их повышенным распадом, при этом процесс разрушения эритроцитов преобладает над процессом регенерации.
Различают две основные группы гемолитических анемий — наследственные и приобретенные. Наследственные гемолитические анемии являются результатом структурных или функциональных генетических дефектов эритроцитов. Каждая из них является самостоятельным заболеванием, имеет специфические клинико-лабораторные признаки, тип наследования, различный прогноз и методы лечения. Приобретенные гемолитические анемии связаны в большинстве случаев с воздействием многочисленных факторов, способствующих повышенному разрушению эритроцитов: антитела, лекарственные препараты (антибиотики, хинин, изониазид, допегит и др.), введение гипотонических растворов, действие высоких и низких температур, ультразвука, гемолитические яды.
Известно, что около 1 % эритроцитов ежедневно удаляются из периферической крови и замещаются равным количеством новых клеток, поступающих из костного мозга. Этот процесс создает в нормальных условиях динамическое равновесие, обеспечивающее постоянное количество эритроцитов в крови. При сокращении продолжительности жизни эритроцитов их разрушение в периферической крови происходит интенсивнее, чем образование в костном мозге и выброс в периферическую кровь. В ответ на сокращение продолжительности жизни эритроцитов, активность костного мозга увеличивается в 6-8 раз, что подтверждается ретикулоцитозом в периферической крови.
Устойчивость эритроцита к различным воздействиям внутренней среды обусловлена структурными белками клеточной мембраны, ее ферментным составом, нормальным гемоглобином и физиологическими свойствами крови и других сред, в которых циркулирует эритроцит. При нарушении свойств эритроцита или изменении среды его пребывания, он преждевременно разрушается в кровеносном русле либо в ретикулогистиоцитарной системе различных органов, прежде всего селезенки. Поэтому в зависимости от локализации принято выделять внутриклеточный и внутрисосудистый варианты гемолиза. В норме наблюдается главным образом внутриклеточный гемолиз, при этом часть эритроцитов ежедневно разрушается, преимущественно в костном мозге и селезенке. При внутриклеточном гемолизе разрушение эритроцитов происходит в клетках ретикулоэндотелиальной системы, прежде всего в селезенке, в меньшей степени в печени, костном мозге.Выделившийся из макрофагов свободный (неконъюгированный) билирубин при попадании в кровоток связывается с альбумином, который доставляет билирубин к гепатоцитам. В печени альбумин отделяется от билирубина, затем в гепатоците происходит связывание неконъюгированного билирубина с глюкуроновой кислотой, при этом образуется моноглюкуронид билирубина (МГБ). МГБ выделяется в желчь, где превращается в диглюкуронид билирубина (ДГБ). ДГБ из желчи выделяется в кишечник, где под влиянием микрофлоры восстанавливается до бесцветного пигмента уробилиногена, и в дальнейшем до пигментированного стеркобилина. При гемолизе резко увеличивается содержание свободного (неконъюгированного, непрямого) билирубина в крови. Гемолиз способствует усиленной экскреции пигментов гема в желчь. Внутриклеточный гемолиз чаще всего сопровождается увеличением печени и селезенки и характерен для наследственных гемолитических анемий. Для лечения этой болезни необходимо определить дефект мембраны эритроцита, из-за которого нарушается иммунный процесс или синтез гемоглобина. При внутрисосудистом гемолизе разрушение эритроцитов происходит непосредственно в кровеносном русле.
Распад эритроцитов внутри сосудов (в крови) приводит к высвобождению гемоглобина, он называется свободный гемоглобин. Этот вид гемолиза характеризуется как раз повышенным содержанием в крови свободного гемоглобина (гемоглобинемия), а в моче повышенным содержанием гемосидерина. Степень гемоглобинемии зависит от интенсивности и скорости распада эритроцитов.
Если количество свободного гемоглобина в плазме превышает резервную гемоглобинсвязывающую емкость гаптоглобина, а поступление гемоглобина из гемолизированных в сосудистом русле эритроцитов продолжается, возникает гемоглобинурия. Появление гемоглобина в моче придает ей темную окраску (цвета темного пива или крепкого раствора перманганата калия). Это обусловлено содержанием как гемоглобина, так и образующегося при стоянии мочи метгемоглобина, а также продуктов распада гемоглобина — гемосидерина и уробилина. Внутрисосудистый гемолиз чаще всего наблюдается при наличии гемолитической анемии, пароксизмальной холодовой агглютининовой болезни, аутоиммунной гемолитической анемии с тепловыми гемолизинами, отравлениях гемолитическими ядами.
Классификация.
I.Наследственные (врожденные) формы гемолитической анемии:
1. Мембранопатии эритроцитов (нарушение строения эритроцитов):
- овалоцитарная,
- акантоцитарная.
- Энзимопенические (ферментопенические) — анемии, связанные с нехваткой какого-либо фермента.
- связанные с дефицитом ферментов глюкозо-6-фосфатдегидрогеназы
- связанныес дефицитом гликолитических ферментов
- связанные с дефицитом ферментов, участвующих в образовании окислении и восстановлении глутатиона
- связанные с дефицитом ферментов, участвующих в метаболизме нуклеотидов
- Гемоглобинопатии:
- серповидно-клеточная анемия
- талассемия
II.Приобретенные формы гемолитической анемии:
- Иммуногемолитические анемии:
- Аутоимунные (вызванные действием антител на антиген эритроцита или нормобластов)
- Изоимунные (гемолитическая болезнь новорожденных, посттрансфузионные)
- Приобретенные мембранопатии вследствие соматической мутации:
- пароксизмальная ночная гемоглобинурия,
- шпороклеточная анемия.
- Связанные с механическим повреждением эритроцитов:
- маршевая гемоглобинурия,
- болезнь Мошкович (микроангиопатическая гемолитическая анемия),
- возникающая при протезировании клапанов сердца
- Токсические.
- гемолитические анемии при приеме лекарственных средств и гемолитических ядов.
III.Другие гемолитические анемии
- гемолитическая желтуха новорожденных, при которой материнские антитела разрушают эритроциты плода или ребенка,
- идиопатическая (примерно 50 % случаев гемолитических анемий)
Клинико-гематологические признаки гемолиза:
Бледно-желтушный цвет кожных покровов, спленомегалия при внутриклеточном гемолизе, в клиническом анализе крови – анемия, ретикулоцитоз, в биохимическом анализе – повышение непрямого билирубина, в моче – увеличение уробилина, в кале – стеркобилина.
Наследственный микросфероцитоз (болезнь Минковского — Шоффара) — одна из форм гемолитической анемии, в основе которой лежит генетический дефект структуры мембраны эритроцитов. Нарушение мембраны ведет к проникновению в эритроцит избытка натрия и воды, вследствие чего образуются сферические эритроциты (сфероциты). Сфероциты, в отличие от нормальных двояковогнутых эритроцитов, не обладают способностью деформироваться в узких участках кровотока, в частности при переходе в синусы селезенки. В результате замедляется их продвижение, отщепляется часть поверхности эритроцита с образованием микросфероцитов. Развивается внутриклеточный гемолиз.
Клиническая картина: первые признаки заболевания выявляются в большинстве случаев в юношеском, реже в зрелом возрасте. В период обострения (гемолитических кризов) возникают слабость, головокружение, может повышаться температура, развивается желтуха, частоспленомегалия, гепатомегалия. Иногда наблюдаются признаки замедленного физического развития больных, а также нарушения лицевого скелета в виде «башенного черепа», седловидного носа, высокого стояния неба, нарушения расположения зубов. В крови выявляется нормохромная анемия, микросфероцитоз, увеличение количества ретикулоцитов. При гемолитических кризах может определяться нейтрофильный лейкоцитоз. В костном мозге отмечается гиперплазия эритроидного ростка. Содержание билирубина в крови повышено за счет непрямой фракции. В моче повышается содержание уробилина, а в кале — стеркобилина. Характерным лабораторным признаком заболевания является снижение осмотической резистентности эритроцитов по отношению к гипотоническим растворам хлористого натрия и повышение кислотной стойкости эритроцитов. При УЗИ могут выявляться камни в желчном пузыре (с наличием клинических проявлений или без них). Важное значение имеет обследование родственников больных, у которых могут выявляться умеренные признаки гемолиза или микросфероциты в крови без клинических проявлений.
Лечение:основным методом лечения больных является спленэктомия, дающая эффект почти во всех случаях. Спленэктомияпоказана при частых гемолитических кризах, резкой анемизации больных, приступах желчнокаменной болезни (одновременно проводится холецистэктомия). При легких компенсированных формах заболевания у взрослых от спленэктомии можно воздержаться.
Дефицит глюкозо-6-фосфатдегидрогеназы (Г6ФДГ)является наиболее распространенной наследственной формой ферментной аномалии эритроцитов, которая может клинически манифестироваться остро возникающими внутрисосудистыми гемолитическими кризами, связанными с приемом медикаментов, инфекциями. Наследование данного ферментного дефицита сцеплено с рецессивной Х-хромосомой, в связи с чем болеют только мужчины. Дефицит Г6ФДГ широко распространен в странах Средиземноморского побережья (Италия, Греция), в Африке, Латинской Америке, Азербайджане, среди некоторых народностей Дагестана, армян и мусульман, проживающих в Иране и Ираке.
К медикаментам, провоцирующим гемолитический криз, относятся противомалярийные препараты (хинин, примахин и др.), производные нитрофурана, изоникотиновой кислоты (изониазид, фтивазид), ПАСК, сульфаниламиды. Некоторые препараты вызывают гемолиз лишь в больших дозах и не оказывают влияния при приеме малых доз (ацетилсалициловая кислота, амидопирин, левомицетин, противодиабетические сульфаниламидные средства). Отмечается лихорадка, головная боль, кожные покровы и склеры бледно-иктеричные, моча меняет цвет (цвет темного пива). При продолжающемся приеме препарата повышается температура, появляются головная боль, одышка, сердцебиение, снижение АД, рвота. Печень часто увеличена и болезненна, селезенка не увеличена. Содержание гемоглобина снижается, иногда значительно (до 20—30 г/л), повышается количество ретикулоцитов. На высоте гемолиза наблюдается лейкоцитоз со сдвигом влево. В эритроцитах, инкубированных с ацетилфенилгидразином, или при специальной окраске выявляются крупные включения (тельца Гейнца). Такие эритроциты могут обнаруживаться и при других ферментодефицитных гемолитических анемиях. В крови определяется повышение содержания непрямого билирубина и свободного гемоглобина. В моче обнаруживается белок (гемоглобинурия) при нормальном осадке. На фоне массивного распада эритроцитов может развиваться синдром диссеминированного внутрисосудистого свертывания крови с нарушениями микроциркуляции в органах и тканях, возникновением острой почечной недостаточности.
Лечение: исключить прием лекарств, провоцирующих гемолиз.
Гемоглобинопатии: талассемия.
Альфа-талассемия распространена в Западной Африке и Южной Азии. Бета-талассемия часто встречается в странах Средиземноморья, Западной Азии и Северной Африки. Это регионы, где распространена малярия. Гетерозиготные носители мутаций в генах альфа- и бета цепей гемоглобина являются более устойчивыми к малярийному плазмодию. Имеются очаги талассемии в Азербайджане, в равнинных районах которого гетерозиготная бета-талассемия наблюдается у 7—10 % населения.
Талассемия – группа наследственных гемолитических анемий, характеризующихся нарушением синтеза полипептидных цепей гемоглобина А.В норме основным вариантом (97 %) гемоглобина взрослого человека является гемоглобин А. Это тетрамер, состоящий из двух мономеров α-цепей и двух мономеров β-цепей. 3 % гемоглобина взрослых представлено гемоглобином А2, состоящем из двух альфа- и двух дельта-цепей. Наличие мутации в генах гемоглобина может привести к нарушению синтеза цепей определённого вида.
Талассемию вызывают точечные мутации или делециив генах гемоглобина, ведущие к нарушению синтеза РНК, что приводит к уменьшению или полному прекращению синтеза одного из видов полипептидных цепей. Синтез цепей другого вида продолжается. Это приводит к образованию нестабильных полипептидных агрегатов из избыточных цепей, нарушающих нормальное функционирование эритроцитов и их разрушению. Повышенный гемолизэритроцитов вызывает анемию.
Причисление талассемии к группе гемоглобинопатии не точно, так как в этом симптомокомплексе по существу не обнаруживается патологического гемоглобина, а лишь гемоглобин плодного (фетального) типа, который, как известно, встречается обычно у здоровых новорожденных. При талассемии образование гемоглобина F продолжается постоянно, не только в грудном возрасте, но и всю жизнь. Дополнительной патологией при гетерозиготной форме талассемии является увеличение количества гемоглобина Hb А2, который в небольшом количестве встречается у здоровых людей. Сущность талассемии заключается в различных нарушениях синтеза гемоглобина: наличии Hb F, торможении образования Hb А и увеличении процентного содержания Hb А2. В зависимости от того, синтез какого из мономеров нарушен, разделяют альфа-, бета- и дельта-талассемию. По тяжести клинических проявлений выделяют тяжёлую, среднюю и лёгкую формы заболевания.
Для талассемии характерны гипохромная анемия,анизоцитоз эритроцитов, наличие мишеневидных форм эритроцитов (пятно гемоглобина в центре клетки, напоминающее мишень). При этом содержание сывороточного железа нормальное или повышенное. Компенсаторная гиперплазия костного мозга, ведёт к нарушениям в строении лицевого черепа. Череп может стать квадратным, башенным; нос приобретает седловидную форму; нарушается прикус и расположение зубов. Отмечается желтушность кожи и слизистых оболочек. Селезёнка и печень увеличены. Больные подвержены инфекционным заболеваниям. Рано начавшаяся анемия обуславливает физическое и умственное недоразвитие ребёнка.
Лечение талассемии. Гетерозиготная форма обычно не требует лечения. При гомозиготной форме, ввиду тяжелой анемии, необходимы периодические переливания крови, без которых дети рано гибнут. Обычно эти переливания производят после 6 месяца жизни, редко раньше.Исключительно в случаях гемолитической анемии, вызванной внеэритроцитарными факторами, осложняющими талассемию, производят спленэктомию. После операции наступает временное улучшение общего состояния, особенно в случаях с увеличенной селезенкой. После спленэктомии наблюдается также увеличение длительности жизни эритроцитов.
Дата добавления: 2017-01-21 ; просмотров: 231 | Нарушение авторских прав
источник
Этиология и патогенез гемоглобинопатий. Клиническая картина талассемии и серповидно-клеточной анемии. Лабораторная и инструментальная диагностика, лечение и профилактика заболеваний крови. Изменения общего анализа крови при патологии эритроцитов.
Студенты, аспиранты, молодые ученые, использующие базу знаний в своей учебе и работе, будут вам очень благодарны.
Размещено на http://www.allbest.ru/
Министерство здравоохранения Республики Беларусь
Белорусский государственный медицинский университет
На тему: «Гемоглобинопатия, талассемия, серповидно-клеточная анемия
Гемоглобинопатия — наследственное или врождённое изменение или нарушение структуры белка гемоглобина, обычно приводящее к клинически или лабораторно наблюдаемым изменениям в его кислород-транспортирующей функции либо в строении и функции эритроцитов.
Гемоглобинопатии подразделяются на две основные категории: серповидно-клеточную анемию и талассемии.
· Серповидно-клеточная анемия характеризуется изменением формы красных кровяных клеток из ровной, кольцевидной в серповидную форму, или форму в виде полумесяца. Такие деформированные клетки теряют пластичность и могут закупоривать мелкие кровеносные сосуды, нарушая кровоток. Это состояние ведет к сокращению срока жизни красных кровяных клеток и последующей анемии, часто называемой серповидно-клеточной анемией. Низкие уровни содержания кислорода в крови и закупорка кровеносных сосудов у людей с серповидно-клеточной анемией могут приводить к синдромам хронической острой боли, тяжелым бактериальным инфекциям и некрозу (отмиранию тканей).
· Талассемии — это тоже наследственные нарушения крови. У людей с талассемией не может вырабатываться достаточно гемоглобина, содержащегося в красных кровяных клетках. Если в красных кровяных клетках недостаточно гемоглобина, кислород не достигает всех частей организма. Органам начинает не хватать кислорода, и они не могут нормально функционировать. Существует два основных типа талассемии — альфа и бета, названные так по двум белковым цепям, из которых состоит нормальный гемоглобин. Как альфа-, так и бета-талассемии имеют легкую и тяжелую формы.
Гемоглобинопатии наследуются от родителей в значительной степени так же, как группа крови, цвет и структура волос, цвет глаз и другие физические черты.
Серповидно-клеточная анемия и тяжелые формы талассемии (большая талассемия) могут появиться только в том случае, если оба родителя являются носителями характерных генов для конкретного состояния. Ребенок, который наследует два таких характерных гена — по одному от каждого родителя — родится с этим заболеванием. Однако шансы ребенка от двух носителей получить два характерных гена и заболеть составляют всего 25%, тогда как шансы стать носителем — 50%. Большинство носителей таких генов ведут совершенно нормальную, здоровую жизнь.
Факты о гемоглобинопатиях
· По оценкам, ежегодно в мире рождается более 300 000 детей с тяжелыми формами этих болезней, причем большинство из них — в странах с низким и средним уровнем дохода.
· Приблизительно 5% населения мира являются здоровыми носителями гена серповидно-клеточной анемии или талассемии. В некоторых регионах доля людей, которые являются носителями этого гена, достигает 25%.
· Такие состояния более всего распространены в тропических районах; однако в результате миграции населения эти болезни распространились в большинство стран.
· Талассемии более всего распространены в Азии, Средиземноморском бассейне и на Ближнем Востоке.
· Серповидно-клеточная анемия преобладает в Африке.
гемоглобинопатия талассемия серповидный анемия
1. Серповидно-клеточная анемия
Серповидно-клеточная анемия является наследственным заболеванием, обусловленным мутацией одного или двух генов, кодирующих образование b-цепей глобина. Данная мутация не возникает в организме больного ребенка, а передается ему от родителей.
В организме человека определяется:
· HbA. Нормальный гемоглобин, состоящий из двух альфа и двух бета-цепей. В норме данная форма составляет более 95% гемоглобина взрослого человека.
· HbA2. Малая фракция, в норме составляющая не более 2% всего гемоглобина взрослого человека. Состоит из двух альфа и двух сигма-цепей глобина.
· HbF (фетальный гемоглобин). Данная форма состоит из двух альфа и двух гамма-цепей и преобладает в период внутриутробного развития плода. Она обладает большим сродством к кислороду, что обеспечивает тканевое дыхание ребенка в период рождения. У взрослого человека доля HbF не превышает 1 — 1,5% и встречается в 1 — 5% эритроцитов.
· HbU (эмбриональный гемоглобин). Начинает образовываться в эритроцитах со 2 недели после зачатия и полностью замещается фетальным гемоглобином после начала кроветворения в печени.
В результате мутации происходит замещение всего лишь одной аминокислоты в структуре b-глобиновой цепи (глутаминовая кислота в 6 позиции заменяется на валин). Это не нарушает процесс образования молекулы гемоглобина в целом, однако приводит к изменению его электрофизиологических свойств. Гемоглобин становится неустойчивым и в условиях гипоксии (недостатка кислорода) изменяет свое строение (кристаллизуется, полимеризуется), превращаясь в гемоглобин S (HbS). Это приводит к изменению формы эритроцита — он удлиняется и истончается, становясь похожим на полумесяц или серп.
Артериальная кровь, оттекающая от легких, насыщена кислородом, поэтому никаких изменений в структуре гемоглобина не происходит. На тканевом уровне молекулы кислорода переходят в клетки различных органов, что приводит к полимеризации гемоглобина и образованию серповидных эритроцитов.
На начальных этапах заболевания данный процесс обратим — при повторном прохождении через легочные капилляры кровь насыщается кислородом, и эритроциты приобретают свою нормальную форму. Однако такие изменения повторяются каждый раз, когда эритроциты проходят через различные ткани и отдают им кислород. В результате этого строение мембраны эритроцитов нарушается, повышается ее проницаемость для различных ионов, что привод к необратимому изменению формы эритроцитов.
Серповидно-клеточная анемия наследуется по аутосомно-рецессивному типу (с неполным доминированием), то есть, чтобы родился больной ребенок, он должен унаследовать мутантные гены от обоих родителей.
Существуют три основные формы серповидно-клеточной анемии и одна дополнительная:
* Серповидно-клеточная анемия (СКА) (HbSS) — пациенты гомозиготны по HbS, то есть фактически весь Hb у них является HbS; у них нет НbА, поскольку нет нормальных генов бета-глобина.
* Серповидно-клеточная болезнь (СКБ) (HbSC) — поражённые дети наследуют HbS от одного из родителей и НbС — от другого (HbC формируется вследствие различных точечных мутаций в бета-глобине), таким образом, у них также отсутствует НbА, поскольку нет нормальных генов бета-глобина.
* Серповидная бета-талассемия — поражённые дети наследуют HbS от одного родителя и признак бета-талассемии — от другого. У них отсутствуют нормальные гены бета-глобина и у большинства пациентов не может формироваться НbА, следовательно, у них имеются симптомы, схожие с симптомами у тех, кто страдает от сереповидно-клеточной анемии.
* Малая анемия (дополнительная форма) — наследование HbS от одного из родителей и гена нормального бета -глобина — от другого, так что, приблизительно 40% Hb является HbS. У них отсутствует серповидно-клеточная анемия (СКА) и, тем не менее, они являются носителями HbS, следовательно, могут передать его потомкам. Они асимптомны и выявляются только по результатам анализов крови.
Талассемимя (анемия Кули) — заболевание, наследуемое по рецессивному типу (двухаллельная система), в основе которого лежит снижение синтеза полипептидных цепей, входящих в структуру нормального гемоглобина. Причины повышенной гибели эритроцитов связаны с нарушенной структурой клетки из-за неправильного соотношения цепей глобина в гемоглобине. Кроме укорочения жизни эритроцитов при данном заболевании происходит гибель клеток предшественников эритроцитов в костном мозге.
Гемоглобин, как известно, состоит из двух видов клеток: гема (пигментные железосодержащие клетки) и гемоглобиновый белок. Этот белок состоит из четырех полипептидных цепочек — две альфа цепочки и две бета цепочки. При нарушениях в синтезе альфа цепочек заболевание именуется альфа-талассемией. Но, самой распространенной формой является именно бета-талассемия, связанная с мутацией в локусе Я-глобина на 11-й паре хромосом, нарушающей синтез Я-глобиновой цепи.
В норме основным вариантом (97 %) гемоглобина взрослого человека является гемоглобин А. Это тетрамер, состоящий из двух мономеров б-цепей и двух мономеров в-цепей. 3 % гемоглобина взрослых представлено гемоглобином А2, состоящем из двух альфа- и двух дельта-цепей. Существуют два гена HBA1 и HBA2, кодирующих мономер альфа, и один HBB-ген, кодирующий мономер бета. Наличие мутации в генах гемоглобина может привести к нарушению синтеза цепей определённого вида.
С талассемией связано более 200 различных мутаций, хотя большинство случаев талассемии вызвано только их небольшим числом. Делеция гена альфа-глобина дает 80-85% случаев альфа-талассемий, а приблизительно 15 мутаций вызывают более 90% случаев — в-талассемий. Молекулярные исследования мутаций как альфа-глобина, так и в-глобина показывают, что различные мутации возникали независимо в разных популяциях, а затем достигали высокой частоты под влиянием отбора.
Причина талассемии — наследственность, вызванная генетическими нарушениями. Но, талассемия у ребенка будет развиваться исключительно, если дефективный ген имеется в наличии у обоих родителей
Чтобы диагностировать анемию серповидно-клеточную необходимо провести следующие исследования: общий клинический анализ крови (при наличии заболевания, в результате будут следующие сведения: сниженное количество здоровых эритроцитов, уровень гемоглобина, гематокрита, и повышенные показатели СОЭ и ретикулоцитов) и специальные пробы, которые определяют наличие серповидных патологических эритроцитов (проба с метабисульфитом натрия(результат положительный) и проба наложения жгута на палец (так же, результат положительный)).
Лабораторная и инструментальная диагностика серповидно-клеточной анемии
1.Общий анализ крови: анемия (НЬ около 80 г/л), цветовой показатель близок к единице, средний эритроцитарный объём > 75 мкм 3 , ретикулоцитоз, лейкоцитоз с нейтрофильным сдвигом лейкоцитарной формулы влево, тромбоцитоз, СОЭ в норме или снижена.
2.Мазок периферической крови: эритроциты в форме серпа (дрепаноциты), мишеневидные эритроциты, полихромазия. Следует отметить, что в обычном мазке крови серповидность не видна. Для её выявления устраняют кислород путём фиксирования мазка парафином с последующей суточной инкубацией или используют «пробу жгута» — накладывают жгут на основание пальца, из которого берут кровь на анализ, создавая таким образом местную выраженную гипоксию.
3.Биохимический анализ крови: повышены концентрация непрямого билирубина и активность ЛДГ в сыворотке крови.
4.Биохимический анализ мочи: обнаруживают уробилин, при тромбозах сосудов почек развивается гематурия.
Пункция костного мозга: гиперплазия эритроидного ростка костного мозга.
Специальный метод исследования: обнаружение полимеризованного HbS при электрофорезе НЬ, повышение осмотической резистентности эритроцитов.
УЗИ: увеличение печени, увеличение (на поздних стадиях — уменьшение) селезёнки, камни жёлчного пузыря.
Подтвердить диагноз можно с помощью электрофареза гемоглобина.
Так же при диагностировании данного вида анемии в анамнезе больного должна присутствовать информация об этнической принадлежности пациента.
Рентгенограмма костей: «волосатая» структура костей черепа, остеопороз трубчатых костей.
Основными методами, применяемыми в процессе диагностики талассемии, являются:
· общий анализ крови (приложение 1);
· биохимический анализ крови (приложение 2);
· дополнительные лабораторные исследования:
1.определение общей железосвязывающей способности плазмы (Нормальные значения ОЖСС находятся в интервале от 45 до 77 мкмоль/л. При талассемии количество свободного железа в крови значительно превышает норму.
2.определение концентрации ферритина в сыворотке крови (При талассемии концентрация железа в крови увеличена, норма у мужчин — 20 — 250 мкг/л, у женщин — 10 — 125 мкг/л.)
3.определение уровня эритропоэтина.(при талассемии данный показатель увеличен в несколько раз, норма 10 — 30 мМЕ/мл)
· рентгенологическое исследование (деформация костей черепа, деформация длинных трубчатых костей, увеличение размеров селезенки и печени)
· ультразвуковое исследование (печень, селезенка, почки и мочевой пузырь)
· пункция костного мозга (при исследовании пунктата костного мозга больных талассемией определяется выраженное увеличение количества клеток, в основном предшественников эритроцитов).
· полимеразная цепная реакция (ПЦР) (можно выявить мутантный ген и хромосому, в которой он расположен, что позволяет подтвердить или опровергнуть диагноз талассемии в 99,9% случаев)
Как для альфа- так и бета-талассемий возможна пренатальная диагностика за счет молекулярного анализа ДНК плода из ворсин хориона или амниоцитов. Молекулярная пренатальная диагностика талассемий наиболее эффективна, если мутации у родителей известны заранее.
Первые признаки заболевания появляются через несколько месяцев после рождения ребёнка, когда уровень HbF снижается. Это связано с тем, что растворимость HbS значительно снижается в условиях гипоксии, а кровь новорождённых насыщена HbF, обладающего большим сродством к кислороду и не имеющего патологической бета-цепи.
Симптомы серповидно-клеточной анемии делятся на две основные категории. Из-за хрупкости красных клеток крови всегда наблюдается анемия, которая может привести к потере сознания, делает больного физически менее выносливым и может вызвать желтуху (связанную с чрезмерным распадом гемоглобина).
Кроме этого, периодическая закупорка мелких капилляров в любой части тела может привести к широкому спектру различных симптомов.
Почти невозможно описать «типичного пациента», страдающего серповидно-клеточной анемией, поскольку симптомы и их тяжесть широко варьируют. Некоторые характерные особенности являются общими почти для всех пациентов с серповидно-клеточной анемией
В периоды гемолитических кризисов отмечается резкое падение уровня гемоглобина, которое сопровождается высокой температурой и черным цветом мочи. У больных серповидной анемией меняется и внешний вид: отмечается высокий рост, худоба, удлиненность туловища, искривление позвоночника, башенный череп и измененные зубы.
Обычно новорождённые вполне здоровы, имеют нормальный вес и нормально развиваются, никаких симптомов у них не проявляется до 3-месячного возраста. Первыми признаками серповидно-клеточной анемии у младенца обычно являются опухание и болезненность кистей рук или стоп, слабость и искривление конечностей и иногда, несколько позднее, отказ от ходьбы. Этот симптом является результатом закупорки эритроцитами капилляров мелких костей кистей и стоп и нарушения кровотока. Эритроциты выпадают из жидкой части крови и откладываются в капиллярах в виде осадка. Скопление эритроцитов постепенно рассасывается само по себе, но до тех пор, пока этого не произойдет, требуется помощь врача, чтобы смягчить боль и обнаружить возможные сопутствующие заболевания. Ребёнок с серповидно-клеточной анемией обычно выглядит бледным, возможно, слегка желтушным, но в остальных отношениях, как правило, здоров.
Единственным очень серьёзным осложнением серповидно-клеточной анемии у ребёнка до 5-летнего возраста является инфекция. Скопление эритроцитов и закупорка капилляров в селезенке, органе, который в норме отфильтровывает бактерии из кровотока, происходит в течение первых лет жизни, что делает ребёнка особенно восприимчивым к смертельному заражению крови — сепсису. Поэтому родителей маленьких детей, страдающих серповидно-клеточной анемией, предупреждают, чтобы они были внимательны и не пропустили ранних симптомов инфекции, таких как раздражительность, нервозность, повышенная температура и плохой аппетит. Родители должны немедленно обращаться за медицинской помощью, если у ребёнка наблюдается какой-либо из этих симптомов. Если при заражении крови достаточно рано начинать применять антибиотики, фатальных осложнений можно избежать. После 5-летнего возраста, когда у ребёнка уже выработались соответствующие естественные антитела к такого рода бактериям, вероятность смертельной бактериальной инфекции существенно снижается.
Проблемой детей школьного возраста с серповидно-клеточной анемией обычно является эпизодическая закупорка эритроцитами капилляров больших костей. В большинстве случаев эти эпизоды протекают относительно легко, наблюдаются лишь слабые ноющие боли в костях.
С возрастом процесс закупорки капилляров может затрагивать и другие органы. Если это произойдет, например, в лёгких, развивается серьёзное респираторное заболевание. Очень редкое осложнение, которое бывает меньше чем у 10% больных с серповидно-клеточной анемией — закупорка сосудов мозга, приводящая к инсульту.
Подростки с серповидно-клеточной анемией испытывают беспокойство и озабоченность из-за того, что их физическое развитие обычно задерживается на 2-3 года. Такие подростки обычно меньше ростом, чем их одноклассники, их часто дразнят за запаздывание в сексуальном развитии. Однако со временем половая зрелость все же наступает, и исследования показывают, что женщины с серповидно-клеточной анемией имеют нормальную возможность к деторождению. Женщины с серповидно-клеточной анемией, безусловно, способны вынашивать и рожать нормальных детей, но во время беременности у них повышается риск осложнений, которые могут привести к выкидышу, преждевременным родам или усилению анемии у матери. Такие беременные женщины должны находиться под наблюдением гинеколога, имеющего специальный опыт по беременности с повышенным риском. В течение беременности таким женщинам может потребоваться переливание крови.
У взрослых с серповидно-клеточной анемией могут обнаруживаться симптомы хронической (постоянной или длительной) закупорки капилляров легких и почек, и может развиться хроническая легочная или почечная недостаточность. Эти два осложнения приводят к ранней смерти некоторых пациентов с серповидно-клеточной анемией.
У других больных может происходить закупорка капилляров сетчатки глаза, что в конечном итоге может привести к слепоте.
Хотя все эти осложнения (почечная и лёгочная недостаточность, слепота, серьёзная инфекция и повторяющиеся костные кризы) характерны для страдающих серповидно-клеточной анемией, крайне редко бывает так, чтобы все они наблюдались у одного пациента.
Так как данное заболевание является наследственным, его симптомы начинают проявляться сразу после рождения либо в первые недели жизни ребенка. Клинические проявления схожи при всех видах талассемии — различается только их выраженность, в зависимости от степени нарушения образования гемоглобина.
Основные проявления талассемии обусловлены:
· нарушением образования эритроцитов (бледность кожных покровов, слабость и повышенная утомляемость, снижение концентрации внимания, замедление роста, снижение аппетита, непереносимость физических нагрузок, язвы кожных покровов, увеличение печени и селезенки.
· избыточным кроветворением в красном костном мозге (при талассемии гиперплазия костного мозга настолько выражена, что происходит деформация костей, в которых он располагается)
· избытком железа в организме (поражение гипофиза, сердца, печени, почек, легких, кожи)
· усиленным разрушением эритроцитов (желтушность кожных покровов, уратовый диатез, увеличение селезенки)
Эффективного лечения серповидно-клеточной анемии, позволяющего раз и навсегда избавиться от данного недуга, на сегодняшний день не существует. Помощь больным заключаются в предотвращении образования большого количества серповидных эритроцитов, а также в устранении симптомов заболевания.
Принципами лечения серповидно-клеточной анемии являются:
Пациентам с серповидно-клеточной анемией рекомендуется:
ь проживать на высоте не более 1500 метров над уровнем моря;
ь проживать в зоне с умеренным климатом (исключающим воздействие экстремально низких или высоких температур);
ь употреблять не менее 1,5 литров жидкости ежедневно;
ь исключить прием алкогольных напитков и наркотиков;
ь отказаться от курения (как самому больному человеку, так и членам его семьи);
ь избегать тяжелых физических нагрузок;
ь выбирать профессию, не связанную с тяжелой физической работой или воздействием высоких/низких температур.
· повышение количества эритроцитов и гемоглобина;
· кислородотерапия (пациент с развивающимся гемолитическим кризом должен быть госпитализирован как можно скорее. Непосредственно в машине скорой помощи ему дается кислородная маска, которая обеспечивает подачу кислорода со скоростью 4 — 6 литров в минуту. После поступления в стационар кислородотерапия продолжается на протяжении нескольких часов или дней)
· устранение болевого синдрома;
· устранение избытка железа в организме;
· профилактика и лечение инфекционных заболеваний.
На сегодняшний день серповидно-клеточная анемия — неизлечимая болезнь.
Профилактика серповидно-клеточной анемии
Как говорилось ранее, серповидно-клеточная анемия неизлечима. Однако современные достижения в области генетики и молекулярной биологии позволяют определить риск рождения детей с данным заболеванием.
Если один или оба родителя больны серповидно-клеточной анемией, то их ребенок также может унаследовать данный недуг. Одним из методов, позволяющим определить вероятность наследования гена, ответственного за развитие данного заболевания, является полимеразная цепная реакция (ПЦР). Суть метода заключается в исследовании генетического материала обоих родителей и выявлении мутантных генов. При этом определяется как их наличие (или отсутствие), так и форма заболевания (гомозиготная или гетерозиготная)
С помощью ПЦР возможно выявить наличие мутантных генов у плода на ранних этапах внутриутробного развития и определить форму заболевания. Это позволяет своевременно поднять вопрос о прерывании беременности.
К сожалению, на современном этапе развития медицины не существует лекарства, способного избавить человека от этого недуга. Большие надежды подает метод пересадки стволовых кроветворных клеток (пересадки костного мозга), однако его выполнение сопряжено с множеством трудностей и не всегда возможно. Вот почему целью лечения в большинстве случаев является устранение симптомов заболевания и предупреждение развития осложнений.
Легкие формы талассемии часто не нуждаются в лечении. Таким пациентам рекомендуется профилактическая сдача общего анализа крови раз в полгода. При более тяжелых формах лечение следует начинать как можно раньше, так как недостаток кислорода в организме может привести к развитию необратимых изменений во внутренних органах.
Основными направлениями в лечении талассемии являются:
· повышение уровня эритроцитов и гемоглобина в крови (единственным эффективным способом повышения количества эритроцитов и гемоглобина в крови является переливание донорской крови. Целевой уровень гемоглобина при этом составляет 100 — 120 г/л)
· устранение переизбытка железа (лечение перегрузки железом следует начинать одновременно с переливанием крови, чтобы предотвратить накопление железа в тканях (в этом случае выведение излишков железа из организма будет более длительным, а поражения органов более выраженными))
· снижение уратового диатеза (с целью снижения количества мочевой кислоты и ее солей в крови назначаются определенные медикаменты, снижающие скорость образования мочевой кислоты, либо способствующие ускоренному выведению ее из организма)
· хирургическое удаление селезенки;
· пересадка костного мозга (суть метода заключается в полном разрушении всего костного мозга пациента и введении в освободившиеся костные полости донорского костного мозга. Если процедура увенчается успехом и донорский костный мозг приживется в организме реципиента, это обеспечит нормальный синтез глобиновых цепей, что устранит центральное звено в развитии талассемии)
Профилактика талассемии основывается на выявлении подверженных риску лиц посредством программ скрининга носителей или изучения историй семьи и предоставления адекватной информации о риске и о возможностях сокращения такого риска. Бета-талассемия обладает уникальным свойством: здоровых носителей можно определять простым, недорогим и точным анализом крови. Таким образом, можно выявлять пары носителей и информировать о генетическом риске до того, как они создадут семью. Большинство пар, подверженных риску талассемии, обращаются за дородовой диагностикой гемоглобинопатии. Стандартный метод диагностики — это взятие пробы ворсинок хорионов и анализ ДНК при сроке беременности 10-12 недель. В большинстве случаев рождение затронутых болезнью детей является результатом неспособности систем здравоохранения адекватно информировать родителей о возможном риске и мерах профилактики, а не тем, что они отвергают тестирование плода. Эффективность служб по борьбе против талассемии зависит от учета их сотрудниками культурной практики и принятия действий, соответствующих данному социальному контексту. При консультировании также необходимо учитывать культурные, религиозные и этические взгляды личности или пары. Успех генетического консультирования в значительной степени определяется его просветительным и добровольным характером.
источник