 Анемия Под определение «гемолитическая анемия» подпадает любая анемия, характеризующаяся преобладанием гемолиза (процессом распада эритроцитов) над эритропоэзом (процессом образования эритроцитов). Гемолиз может быть иммунный и неиммунный. Гемолитическая анемия бывает наследственные и приобретенные.
Анемия Под определение «гемолитическая анемия» подпадает любая анемия, характеризующаяся преобладанием гемолиза (процессом распада эритроцитов) над эритропоэзом (процессом образования эритроцитов). Гемолиз может быть иммунный и неиммунный. Гемолитическая анемия бывает наследственные и приобретенные.
- Наследственная форма гемолитической анемии, обусловленная нарушением мембраны эритроцитов
- Наследственная форма гемолитической анемии, обусловленная нарушением активности ферментов эритроцитов
- Наследственная форма гемолитической анемии, обусловленная нарушением синтеза или структуры гемоглобина
- Анемия, обусловленная влиянием антител
- Анемия, обусловленная изменением структуры мембраны, вызванной соматической мутацией
- Анемия, обусловленная механическим повреждением оболочки эритроцитов
- Анемия, вызванная химическим повреждением эритроцитов
- Анемия, вызванная дефицитом витаминов (фолиевой кислоты и цианокобаламина)
- Анемия, вызванная разрушением эритроцитов паразитами
Болезнь Минковского-Шоффара (наследственный микросфероцитоз) – группа наследственных гемолитических анемий, характеризующихся образованием микросфероцитов (шаровидных эритроцитов) и обусловленных дефектом протеинов цитоскелета эритроцитов. При этом эритроциты теряют часть мембраны, уменьшается соотношение площади поверхности к объему, в результате чего эритроцит превращается в микросфероцит. Как правило, патология наследуется по аутосомно-доминантному признаку. Распространенность наследственного микросфероцитоза составляет примерно 1 случай на 1000-4500 человек.
При наследственном микросфероцитозе генетические нарушения влияют на протеины цитоскелета, преимущественно на те, которые объединяют цитоскелет с мембраной эритроцита. У большинства больных отмечается значительный дефицит спектрина, и только в некоторых случаях этот дефицит обусловлен генетическими дефектами самого спектрина.
Главные признаки наследственного микросфероцитоза – анемия, желтуха, спленомегалия (увеличенная селезенка). Анемия возникает из-за внутриклеточного распада эритроцитов. Желтуха развивается посредством непрямой гипербилирубинемии, может быть непостоянной и, как правило, слабо выражена у детей раннего возраста. Повышенное содержание билирубина в желчи часто является причиной образования пигментных желчных камней (даже у детей). Увеличение селезенки (спленомегалия) отмечается практически во всех случаях. При системных инфекционных патологиях интенсивность гемолиза может увеличиваться, в результате чего развивается спленомегалия.
Тяжелые формы наследственного микросфероцитоза характеризуются деформацией скелета: изменение расположения зубов, акрокефалия (башенный череп), высокое верхнее небо, микрофтальмия (уменьшение глазного яблока). В некоторых случаях отмечаются укороченные мизинцы. Могут образовываться трофические язвы на ногах.
Наследственный микросфероцитоз сопровождается апластическими кризами, которые провоцируются инфекцией (особенно парвовирусной).
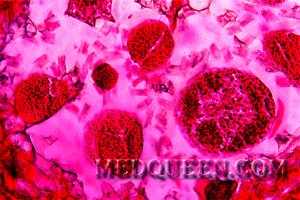
Микросфероцитоз – характерное изменение формы эритроцитов при этой патологии. При анализе мазка крови в биологическом материале наблюдаются микросфероциты в виде мелких клеток без центрального просветления (см рисунок 1). Отметим, что обнаружение микросфероцитов в мазках не всегда является признаком наследственного сфероцитоза.
Рисунок 1. Наследственный микросфероцитоз. Микросфероциты в мазке периферической крови (окр. по Романовскому-Гимзе, ув. ×100)
Такой признак обнаруживается при аутоиммунной гемолитической анемии с неполными тепловыми агглютинами, при наследственных дизэритропоэтической анемии. Средний объем эритроцитов, как правило, остается в норме или незначительно снижен. Показатель среднего содержания гемоглобина в эритроцитах в норме или незначительно повышен. Средняя концентрация гемоглобина в эритроцитах повышена почти у 50% пациентов.
Количественным показателем сферичности эритроцитов является осмотическая устойчивость (она снижена). Уровень ретикулоцитов в крови при гемолитическом кризе может значительно повышаться. Миелограмма показывает резкое раздражение красного ростка. Дифференциальный диагноз проводят с аутоиммунной гемолитической анемией, для которой характерна положительная проба Кумбса, отсутствие этой патологии среди родственников пациента и отсутствие данных о начале заболевания в детском возрасте.
Основной метод лечения анемии при наследственном микросфероцитозе – спленэктомия, с помощью которой устраняется анемия; при этом нельзя устранить морфологический дефект эритроцитов.
Наследственная гемолитическая анемия, обусловленная дефицитом глюкозо-6-фосфат дегидрогеназы эритроцитов – наиболее распространенная ферментопатия эритроцитов из группы ферментопатий пентозофосфатного пути метаболизма глюкозы. Глюкозо-6-фосфатдегидрогеназа эритроцитов – олигомер (в зависимости от условий может быть димер или тетрамер), который состоит из субъединиц с молекулярной массой 56 000 D. По данным ВОЗ (Всемирной организации здравоохранения) во всем мире количество людей, страдающих этой патологией, составляет более 200 млн. Наиболее широкое распространение этого заболевания характерно для Средиземноморского региона (Сицилия, Греция, Сардиния), негроидной расы, жителей Ближнего и Дальнего востока.
Клиническая картина при наследственной форме гемолитической анемии полиморфна: степень тяжести патологии может колебаться от гемолитической анемии, возникающей спонтанно после рождения, до гемолитических кризов. Гемолитический криз, который может провоцироваться метаболическим ацидозом или гипогликемией, развивается за несколько часов. В тяжелых случаях у больного развивается гемоглобинурия и шок. Также наблюдаются желтуха, моча приобретает бурый или черный цвет, одышка, диарея, рвота, снижение артериального давления, развивается тяжелая анемия, увеличиваются печень (гепатомегалия) и селезенка (спленомегалия).
Тяжелый гемолитический криз может спровоцировать развитие ДВС-синдрома (диссеминированного внутрисосудистого свертывания крови). Некоторые пациенты не переносят конские бобы (Viciafaba), после употребления которых происходит молниеносное развитие гемолитического криза (это явление также известно, как фовизм или примахиновая анемия).
Дефицит глюкозо-6-фосфат дегидрогеназы эритроцитов необходимо подозревать во всех случаях острого гемолиза, особенно у лиц негроидной расы и жителей средиземноморского региона. Диагноз подтверждается путем проведения лабораторных анализов. Острый гемолиз характеризуется быстрым снижением гематокрита с одновременным повышением уровня гемоглобина и непрямого гемоглобина, а также снижением уровня гаптоглобина. Анализ мазка крови показывает наличие фрагментов эритроцитов. Основой диагностики считается качественное (при необходимости – количественное) определение активности глюкозо-6-фосфат дегидрогеназы эритроцитов. У пациентов с вариантом «А-» явление аномального гемолиза проходит, как правило, самостоятельно – такие больные не нуждаются в специальном лечении. В случае развития тяжелого гемолитического криза необходимо проводить форсированный диурез, профилактику ДВС-синдрома, плазмаферез (с целью удаления продуктов гемолиза).
В случае возникновения качественной гемоглобинопатии происходит изменение аминокислотной последовательности цепей глобина. Талассемия (количественная гемоглобинопатия) характеризуется снижением образования цепей глобина без изменения их цепей. Нужно отметить, что разница между качественной и количественной гемоглобинопатиями не абсолютна.
Талассемия (анемия Кули) – группа патологий, обусловленных генетическим нарушением синтеза одной из цепей глобина. В норме процесс синтеза глобиновых цепей сбалансирован, поэтому свободных цепей глобина нет. В случае нарушения синтеза одной из цепей глобина баланс нарушается, образуются лишние цепи, которые агрегируют и откладываются в эритрокариоцитах. Среди жителей Средиземноморья наиболее распространена β-талассемия.
«Большая талассемия» (болезнь Кули, β-талассемия) – наследственная гемолитическая анемия, впервые описанная американскими педиатрами-гематологами Томасом Бентоном Кули (Thomas Benton Cooley) и Ли (P. Lee) в статье «Серия случаев спленомегалии у детей с анемией и необычными изменениями костей» («A Series of Cases of Splenomegaly in Children, with Anemia and Peculiar Bone Changes»), где были приведены случаи у выходцев из стран Средиземноморья. Для анемии Кули характерна тяжелая степень течения с самого детства, задержка роста и изменения костей в результате увеличения объема костного мозга, возникающие в случае отсутствия соответствующего лечения). Также при этой патологии у больного наблюдаются гепатомегалия, спленомегалия, гиперспленизм, деформации черепа (монголоидное лицо, башенный череп); желтуха, бледность и отложение меланина придают коже особый медный оттенок. Кроме этого, наблюдается перегрузка железом сердца, легких, печени, поджелудочной железы и других органов эндокринной системы, переломы костей, сдавления периферических нервов, разного рода инфекционные осложнения.
Результаты лабораторных исследований периферической крови показывают гипохромную анемию, ретикулоцитоз, мишеневидные эритроциты (см рис 2-4).
Рисунок 02. Анемия Кули (большая талассемия). Периферическая кровь. Микроцитоз, выраженная гипохромия, мишеневидные нормобласты и эритроциты (окр. по Романовскому-Гимзе, ув. ×100)
Рисунок 03. Анемия Кули (большая талассемия). Периферическая кровь (окр. по Романовскому-Гимзе, ув. ×50)
Рисунок 04. Анемия Кули (большая талассемия). Периферическая кровь. Множественные мишеневидные эритроциты (окр. по Романовскому-Гимзе, ув. ×100)
Миелограмма демонстрирует раздражение «красного ростка» и повышение количества сидеробластов. Также наблюдается повышение осмотической резистентности эритроцитов и количества билирубина за счет непрямой фракции. В крови повышается содержание железа и ферритина, развивается гемосидероз (чрезмерное отложение гемосидерина в тканях) внутренних органов. При гомозиготной β-талассемии необходимо проводить пренатальную диагностику – забор клеток плода из амниотической жидкости на предмет выявления мутации генов, отвечающих за кодирование β-цепи глобина, с применением метода полимеразной цепной реакции.
Без соответствующего лечения больные анемией Кули умирают в детском возрасте. Продлить жизнь, предупредить деформации костей и задержку роста можно путем регулярных трансфузий эритроцитарной массы (лучше переливать отмытые или размороженные эритроциты) при условии поддержания достаточно высокого уровня гемоглобина. В случае значительной спленомегалии и явлениях гиперспленизма больному показана спленэктомия (удаление селезенки). С целью предотвращения развития гемосидероза пациентам периодически назначают Деферазирокс (Эксиджад) или Дефероксамин (Десферал). Излечение возможно при аллогенной трансплантации костного мозга.
Серповидноклеточная анемия обусловлена носительством гемоглобина, который меняет свою структуру в условиях гипоксии. Самой распространенной аномалией структуры гемоглобина является гемоглобинопатия Sα2β26 глу+вал. При гомозиготном носительстве можно говорить о серповидноклеточной анемии; при гетерозиготном носительстве – серповидноклеточная аномалия. Патология наследуется по аутосомно-доминантному признаку. При серповидноклеточной анемии наблюдается мутация, в результате которой в цепи глобина глутаминовая кислота заменяется валином. В результате растворимость гемоглобина S при отдаче кислорода снижается, что приводит к образованию геля.
Серповидноклеточная анемия наиболее распространена среди населения Центральной Африки, Турции, Индии, Кубы. У больных диагностируется анемия, тромботические осложнения, поражения костей и суставов (отмечаются некрозы плечевой и бедренной костей). Кроме этого, тромбозы осложняются инфарктами (сердца, легких, почек, селезенки, головного мозга), приступами сильной боли в области живота. У детей отмечаются нарушения физического (отставание в росте) и полового развития, ночное недержание мочи, нарушение зрения (тромбозы сосудов сетчатки). Также могут развиваться гемолитический, апластический и секвестрационные кризы, при этом в селезенке происходит резкое накопление эритроцитов, что вызывает гиповолемический шок и резкое снижение уровня гемоглобина.
Для анализов крови при апластической анемии характерны низкий уровень гемоглобина, наличие серповидных эритроцитов (рисунок 5), базофильная пунктация эритроцитов, их мишеневидность, повышение уровня ретикулоцитов и непрямого билирубина. Миелограмма демонстрирует раздражение «красного ростка».
Рисунок 5. Серповидноклеточная анемия. Периферическая кровь. Серповидные и мишеневидные эритроциты. выраженная гипохромия эритроцитов (окр. по Романовскому-Гимзе, ув. ×100)
В качестве лечения применяют адекватную инфузионную терапию, переливания эритроцитарной массы, оксигенотерапии.
К приобретенным гемолитическим анемиям относится группа заболеваний разного патогенеза, которые объединяет внутрисосудистый гемолиз (гемолиз эритроцитов в периферической крови). В зависимости от механизма эритролиза приобретенная гемолитическая анемия может носить иммунный и неиммунный характер. Но, несмотря на разные патогенетические механизмы, клинические признаки этих анемий часто совпадают.
Гемолитическая анемия у пациентов с протезированными клапанами сердца и сосудами развивается примерно в 10% случаев при протезированном аортальном клапане. При использовании стеллитовых запирательных элементов частота гемолиза незначительно увеличивается (по сравнению с селиконовыми). Также некоторое увеличение частоты гемолиза отмечается при наличии околоклапанной регургитации и при малом диаметре клапана. Биопротезы (свиные клапаны) в редких случаях являются причиной механического гемолиза. Гораздо реже причиной гемолиза может быть также протезированный митральный клапан, так как трансклапанный градиент давления в этом случае ниже.
Гемолиз протезированными клапанами происходит в результате одновременного действия сразу нескольких факторов:
- Значительная сила сдвига, которая при турбулентном токе крови действует на мембрану эритроцитов, особенно когда под высоким давлением кровь проходит через маленькое отверстие (например, при околоклапанной регургитации)
- Отложения фибрина на участках неплотного прилегания кольца клапана к тканям сердца
- Прямое механическое повреждение эритроцитов при закрытии запирательного элемента
Значительное разрушение эритроцитов может наблюдаться после закрытия дефекта межпредсердной перегородки типа ostium primum заплатой из синтетического материала. Умеренное сокращение жизни эритроцитов с легкой анемией или без нее может наблюдаться при значительном обызвествлении аортального клапана. Механический гемолиз обнаруживается также у пациентов, перенесших аортокоронарное и аортобедренное шунтирование.
Тяжелые случаи механического гемолиза сопровождаются тяжелой анемией, ретикулоцитозом, обнаруживаются фрагментированные эритроциты (шизоциты), гемоглобинемия и гемоглобинурия, повышается активность лактатдегидрогеназы, снижается уровень гаптоглобина. Выведение железа из организма с мочой в виде гемосидерина или гемоглобина может вызвать дефицит железа в организме. В случае развития дефицита железа пациенту назначается пероральный прием препаратов железа. Терапия препаратами железа способствует повышению уровня гемоглобина и способствует снижению сердечного выброса и снижению интенсивности гемолиза. Отметим, что ограничение физической активности также способствуют снижению интенсивности распада эритроцитов. Если предпринимаемые меры не приводят к желаемому результату, нужно полностью устранить околоклапанную регургитацию или заменить протез.
источник
приобретенные
| Токсическая гемолитические яды, соединения мышьяка, свинца; возбудители инфекций: гемолитический стрептококк, малярийный плазмодий) | Иммунная (перелива-ние несов-местимой крови; Rh-несов-местимость крови матери и плода | Механическая (механическое повреждение эритроцитов при протезировании сосудов и клапанов сердца | Приобретенная (соматические мутации при образовании аутоантител против собственных эритроцитов под влиянием вирусов и лекарств с образованием патологических популяций эритроцитов с нарушенной мембраной |
Наследственная гемолитическая анемия— наследственный микросфероцитоз или болезнь Минковского-Шоффара. Неполноценность эритроцитов при болезни Минковского-Шоффара обусловлена генуинной недостаточностью синтеза АТФ, необходимого для поддержания двояковогнутой формы эритроцитов. При снижении содержания АТФ ниже 10 % от нормы эритроциты теряют ионы калия, в них поступает избыточное количество ионов Nа + и воды, эритроциты изменяют свою форму и превращаются в сфероциты. Кроме того, понижается их осмотическая резистентность, что связано с уменьшением содержания в мембране актомиозиноподобного белка, падением количества фосфолипидов и холестерина. Эритроциты живут 2-3 недели. Селезенка увеличена у всех больных, а печень — у половины пациентов.
Картина крови при наследственных гемолитических анемиях. Отмечается усиленная регенерация эритроцитарного ростка. При частых гемолитических кризисах может быть регенераторная анемия. В мазке крови, наряду с регенеративными формами (высокий ретикулоцитоз — до 50-60 %, полихроматофилия, ядерные формы эритроцитов), находится дегенеративно измененные эритроциты малого размера —
Выделяют три клинические формы:
а) врожденная анемия новорожденных — анемическая форма,
б) желтуха новорожденных — желтушная форма,
в) врожденная водянка — отечная форма.
Развитие той или иной формы гемолитической болезни зависит от типа и титра антител. Тяжелые формы (отечная и желтушная) зависят от титра неполных антител, а анемическая — от агглютинирующих (полных) антител. Резус-конфликт наблюдается у 2-3 на 1000 новорожденных, а по системе АВО у 5-6 на 1000.
Патогенез. При первой беременности возможна иммунизация матери, но эта беременность обычно протекает безопасно. Последующие беременности протекают под угрозой вторичного иммунного ответа, когда появляются иммуноглобулины IgG, более мощные, более реактивные. Они легко проникают через плаценту в кровь плода и разрушают его эритроциты. Картина крови нестабильна. Очень много биллирубина. Повреждаются ядра нервных клеток головного мозга.
Отечная формахарактеризуется общими выраженными отеками, кожа плода бледная, полупрозрачная, в полостях тела — транссудат. Сердце, печень, селезенка значительно увеличены, почки полнокровны. Мягкая мозговая оболочка и ткань мозга отечны и полнокровны.
При анемической формевыражены общая бледность покровов и малокровие внутренних органов. В печени и селезенке — умеренный эритробластоз. Изменения головного мозга менее выражены, чем при отечной форме.
Желтушная формахарактеризуется желтухой покровов, подкожно-жирового слоя и интимы крупных сосудов. Количество непрямого биллирубина в сыворотке крови достигает 20-40 мг %. Ядра головного мозга прокрашены в охряно-желтый цвет. Тяжесть поражения усугубляется гипоксией из-за повреждений мелких сосудов.
Аналогично, но менее выраженно, протекает гемолитическая желтуха при несовместимости групп крови плода и матери.
Картина крови при приобретенных гемолитических анемиях. Приобретенная гемолитическая анемия может быть по типу кроветворения нормобластической, по регенераторной способности костного мозга — регенераторной, по цветовому показателю — нормо- или гиперхромной.
Степень уменьшения количества эритроцитов и гемоглобина зависит от интенсивности гемолиза. В мазке крови обнаруживают клетки физиологической регенерации и дегенеративно измененные эритроциты (пойкилоцитоз, анизоцитоз). Появление большого количества эритробластов и нормобластов характерно для гемолитической болезни новорожденных. Профилактика — обменное переливание крови отца или введение матери антирезусных антител.
Аутоиммунные гемолитические анемиивозникают при изменении антигенной структуры мембраны эритроцитов при переохлаждении, ожогах, инфекциях и действии лекарственных препаратов. В патогенезе главным является разрушение эритроцитов, образование аутоантител, стимуляция иммунного ответа и образуется порочный круг (самая опасная анемия). Лечение этого процесса — подавление иммунного ответа гормонами и иммунодепрессантами.
Патогенез и гематологическая характеристика. Гемолиз эритроцитов происходит внутри сосудов с образованием непрямого биллирубина. Развивается желтуха. Цветовой показатель может быть в норме или повышен. Размер эритроцитов — нормоциты и умеренный макроцитоз, пойкилоцитоз. Повышена регенерация эритроцитов, гипер- или нормохромия, повышена регенерация эритроцитов.
Анемии вследствие нарушения кровообразования. Витамин В12— и фолиеводефицитные анемии- это анемии, связанные с нарушением синтеза нуклеиновых кислот и заменой нормобластического типа кроветворения мегалобластическим из-за недостатка в организме витамина В12 и фолиевой кислоты.
В патогенезевитаминВ12-и-фолиеводефицитной анемии различают 3 основные ветви:
а) нарушение костномозгового кроветворения (что ведет к гипоксии);
б) нарушение миелинизации нервных волокон (нарушения чувствительности от парестезий до синдрома демиелинизации);
в) желудочно-кишечные (нарушение образования сосочков языка — глоссит, патология пристеночного пищеварения).
Дефицит витамина В12 и фолиевой кислоты, участвующих в образовании тимина, входящего в состав ДНК, снижает скорость ее образования. Нарушение клеточного деления приводит к формированию крупных клеток крови: мегалоцитов, мегалобластов, гигантских мегакариоцитов. Созревание мегалобластов до мегалоцитов сопровождается нарушением энуклеации — об этом свидетельствуют появление в мегалоцитах телец Жолли (остатки ядра) и колец Кебота (остатки ядерной оболочки). Наличие большого количества мегалобластов и мегалоцитов, насыщенных гемоглобином, обуславливает гиперхромию (ЦП>1,5).
Картина крови. Витамин В12-и-фолиеводефицитная анемия — это анемия мегалобластическая, гиперхромная, диспластическая, макроцитарная. В мазке крови появляются мегалобласты — клетки патологической регенерации костного мозга и мегалоциты (крупные клетки с базофильной, полихроматофильной или оксифильной цитоплазмой, для которых характерна ранняя гемоглобинизация). В крови встречается много дегенеративно измененных эритроцитов: пойкилоцитоз, анизоцитоз с микроцитозом, гипохромные эритроциты, мегалоциты с патологическими включениями. Уменьшается количество клеток физиологической регенерации (ретикулоциты, полихроматофилы), т.к. в костном мозге наблюдается раздражение эритроцитарного ростка с преобладанием мегалобластического типа кроветворения над нормобластическим. Наблюдается тромбоцитопения и лейкоцитопения с атипическими клетками.
Железодефицитная анемия (сидеропеническая) — это анемия, вызванная недостатком железа в организме в результате нарушения баланса между его поступлением, потреблением и потерей. Это самый распространенный вид анемии (80-90 % всех анемий). Недостаток железа в организме проявляется исчезновением гемосидерина в клетках печени и селезенки, снижением количества сидеробластов и сидероцитов в костном мозге. В крови уменьшается содержание сывороточного железа и степень насыщения им трансферрина (белка-переносчика железа), что ведет к снижению транспорта железа в костный мозг. Нарушается включение железа в эритроцитарные клетки, при этом снижается синтез гема и глобина, уменьшается активность некоторых ферментов в эритроцитах, что вызывает повышение их чувствительности к окислителям (т.к. неполноценность ферментативных процессов ведет к неустойчивости клеточных мембран), эритроциты подвергаются гемолизу под действием окислителей и продолжительность жизни эритроцитов уменьшается.
Картина крови. Железодефицитная анемия — это нормобластическая, гипохромная анемия (из-за недостаточной гемоглобинизации). В мазке крови наблюдается анизоцитоз (микроцитоз), пойкилоцитоз, шизоцитоз — обломки эритроцитов. Количество ретикулоцитов зависит от регенерирующей способности костного мозга (анемия может быть сначала регенераторной, а затем гипорегенераторной).
Железорефрактерная анемия —(рефрактерный — невосприимчивый)- сидероахрестическая, сидеробластическая, железонасыщенная анемия — обширная группа анемических состояний с высоким содержанием сывороточного железа, костномозговым сидеробластозом (отложение органических и неорганических соединений железа) и рефрактерностью к лечению железом. Анемии могут быть наследственными и приобретенными.
Этиология и патогенез связаны с нарушением активности ферментных систем, участвующих в биосинтезе гема. Наследственные анемии встречаются почти исключительно у мальчиков и молодых мужчин, приобретенные первичные формы чаще у пожилых лиц обоего пола, а вторичные — в любом возрасте.
Картина крови: выраженная анемия (возможно снижение гемоглобина до 3 г%, эритроцитов до 1 млн в микролитре). Процент ретикулоцитов в пределах нормы. Величина эритроцитов в норме или несколько завышена за счет макроцитов. Нередко отмечается клеточный диморфизм с наличием двух популяций эритроцитов: гипохромно-микроцитарной и нормохромно-макроцитарной. Лечение и прогноз зависит от формы анемии.
источник
Анемия, при которой процесс разрушения эритроцитов преобладает над процессом регенерации, называется гемолитической.
Естественная гибель эритроцита (эритродиерез) происходит спустя 90-120 дней после его рождения в сосудистых пространствах ретикулогистиоцитарной системы, главным образом в синусоидах селезенки и значительно реже непосредственно в кровеносном русле. При гемолитической анемии наблюдается преждевременное разрушение (гемолиз) эритроцитов. Устойчивость эритроцита к различным воздействиям внутренней среды обусловлена как структурными белками клеточной мембраны (спектрин, анкирин, белок 4,1 и др.), так и ее ферментным составом, кроме того, нормальным гемоглобином и физиологическими свойствами крови и других сред, в которых циркулирует эритроцит. При нарушении свойств эритроцита или изменении среды его пребывания, он преждевременно разрушается в кровеносном русле либо в ретикулогистиоцитарной системе различных органов, прежде всего селезенки.
Гемолитические анемии гетерогенны по своему патогенезу, поэтому установление механизма гемолиза является важной клинической задачей, не всегда легко решаемой.
Обычно выделяют наследственные и приобретенные гемолитические анемии, поскольку они имеют различные механизмы развития и отличаются подходом к лечению. Реже классифицируют гемолитические анемии по наличию или отсутствию иммунопатологии, различая аутоиммунные и неиммунные гемолитические анемии, к которым относятся врожденные гемолитические анемии, приобретенные гемолитические анемии у больных циррозом печени, а также при наличии протезов сердечных клапанов и так называемая маршевая гемоглобинурия.
Гемолитическим анемиям присущ ряд признаков, которые выделяют их из анемий другого происхождения. Прежде всего, это гиперрегенераторные анемии, протекающие с гемолитической желтухой и спленомегалией. Высокий ретикулоцитоз при гемолитическим анемиям обусловлен тем, что при распаде эритроцитов образуются все необходимые элементы для построения нового эритроцита и, как правило, отсутствует де фицит эритропоэтина, витамина В12, фолиевой кислоты и железа. Разрушение эритроцитов сопровождается увеличением содержания в крови свободного билирубина; когда его уровень превышает 25 мкмоль/л, появляется истеричность склер и кожных покровов. Увеличение селезенки (спленомегалия) — результат гиперплазии ее ретикулогистиоцитарной ткани, обусловленной повышенным гемолизом эритроцитов. Общепринятой классификации гемолитических анемий нет.
Наследственные гемолитические анемии.
А. Мембранопатии вследствие нарушения структуры белка мембраны эритроцита:
Микросфероцитоз; эллиптоцитоз; стоматоцитоз; пиропойкилоцитоз
Нарушение липидов мембраны эритроцитов: акантоцитоз, дефицит активности лецитин-холестерин-ацилтрансферазы, увеличение содержания лецитина в мембране эритроцитов, детский инфантильный пикноцитоз
Дефицит ферментов пентозофосфатного цикла
Дефицит активности ферментов гликолиза
Дефицит активности ферментов обмена глутатиона
Дефицит активности ферментов, участвующих в использовании АТФ
Дефицит активности рибофосфатпирофосфаткиназы
Нарушение активности ферментов, участвующих в синтезе порфиринов
Обусловленные аномалией первичной структуры гемоглобина
Вызванные снижением синтеза полипептидных цепей, входящих в состав нормального гемоглобина
Обусловленные двойным гетерозиготным состоянием
Аномалии гемоглобина, не сопровождающиеся развитием заболевания
Приобретенные гемолитические анемии
А. Иммунные гемолитические анемии:
Гемолитические анемии, связанные с воздействием антител: изоиммунные, гетероиммунные, трансиммунные
Аутоиммунные гемолитические анемии: с неполными тепловыми агглютининами, с тепловыми гемолизинами, с полными Холодовыми агглютининами, связанные с двухфазными холодовыми гемолизинами
Аутоиммунные гемолитические анемии с антителами против антигена нормоцитов костного мозга
Б. Гемолитические анемии, связанные с изменением мембран, обусловленные соматической мутацией: ПНГ
В. Гемолитические анемии, связанные с механическим повреждением оболочки эритроцитов
Г. Гемолитические анемии, связанные с химическим повреждением эритроцитов (свинец, кислоты, яды, алкоголь)
Д. Гемолитические анемии на фоне дефицита витаминов Е и А
Е. Гемолитические анемии, обусловленные разрушением эритроцитов паразитами (малярия)
На этапе клинического анализа крови врач-лаборант изучает морфологию эритроцитов. При этом могут быть выявлены различные ее изменения: микро-сферо-, овало-, эллипто-, стомато-, аканто-, пиропикноцитоз, мишеневидность эритроцитов, что дает основание предположить один из вариантов мембранопатии, а мишеневидность эритроцитов характерна для талассемии. При наличии в эритроцитах телец Гейнца-Эрлиха на фоне анизопойкилоцитоза можно предположить один из вариантов наследственной ферментопатии. При серповидноклеточной гемолитической анемии проводят метабисульфитную пробу или пробу с герметизацией капли крови, что позволяет увеличить число серповидных эритроцитов и тем самым облегчает диагностику. Внутрисосудистый гемолиз проявляется наличием фрагментированных эритроцитов, количество которых иногда достигает 100 %, что наблюдается при ДВС-синдроме, сопровождающем многие тяжелые заболевания, а также при отравлении гемолитическими ядами, маршевом гемолизе и при искусственном клапане сердца. Таким образом, измененная морфология эритроцитов, характерная для тех или иных вариантов гемолитической анемии, позволяет обосновать дальнейший диагностический поиск.
Уже при первом знакомстве с больным анемией целесообразно выяснить его принадлежность к той или иной этнической группе, поскольку известно, что азербайджанцы, жители, населяющие Дагестан, грузины и горские евреи чаще болеют наследственной гемолитической анемией. Следует расспросить больного, нет ли среди его кровных родственников больных с анемией, когда у него появились первые симптомы анемии, когда впервые был установлен диагноз анемии. О наследственном характере гемолитической анемии иногда свидетельствует наличие желчнокаменной болезни, диагностированной у больного или его родственников в молодом возрасте (гипербилирубинемия может способствовать камнеобразованию в желчном пузыре и протоках).
При физикальном обследовании больных с наследственной гемолитической анемией в части случаев выявляют изменения костного скелета, строения черепа. Совокупность данных анамнеза, результатов физикального и лабораторного исследований позволяет определить гемолитический характер анемии. Дальнейшее исследование направлено на уточнение основного патогенетического звена гемолитической анемии.
Существуют клинические и лабораторные различия при внутрисосудистом и внутриклеточном гемолизе. Так, при разрушении эритроцитов в селезенке, печени, костном мозге в макрофагах происходит катаболизм гема: под влиянием фермента гемоксигеназы образуется вердогемоглобин, отщепляется железо, затем образуется биливердин, который под влиянием биливердинредуктазы превращается в билирубин. Попадая в общий кровоток билирубин связывается с альбумином; в печени альбумин отщепляется, а билирубин соединяется с глюкуроновой кислотой, образуя моно- и диглюкуронид билирубина, которые поступают в желчь и выделяются в кишечник. Там под влиянием микрофлоры он превращается в уробилиноген, а затем — в стеркобилин. Этот процесс аналогичен физиологическому: примерно 1 % эритроцитов ежедневно погибает преимущественно в ретикулогистиоцитарной системе селезенки, печени, костного мозга. Но при гемолитической анемии гемолиз резко нарастает, поэтому в крови увеличивается содержание свободного билирубина, усиливается его экскреция в желчь, нарушая ее коллоидную стабильность, и создаются предпосылки к развитию холелитиаза.
Часть эритроцитов разрушается в кровеносном русле и в норме. При этом свободный гемоглобин связывается с плазменными белками: гаптоглобином, гемопексином, альбумином. Образовавшиеся комплексы захватываются гепатоцитами, а затем удаляются клетками ретикулогистиоцитарной системы. Если разрушение эритроцитов происходит непосредственно в кровеносном русле, а количество свободного билирубина превышает гемоглобинсвязывающую емкость гаптоглобина, то свободный гемоглобин проникает из крови в мочу через гломерулярный барьер почек: возникает гемоглобинурия, и моча приобретает темную окраску.
Ценным показателем гемолиза является уровень гаптоглобина: чем интенсивнее гемолиз, тем больше расходуется гаптоглобина; при этом его расход превышает синтетическую способность печени (гаптоглобин синтезируется в печени, принадлежит к классу а2-глобулинов), в связи с чем уровень гаптоглобина резко снижается, что наблюдается прежде всего при внутрисосудистом гемолизе.
Таким образом, клиническими и лабораторными признаками, характерными для внутриклеточного гемолиза, являются: желтушность кожи и склер, спленомегалия, увеличенное содержание свободного билирубина, снижение уровня гаптоглобина. Для внутрисосудистого гемолиза увеличение селезенки нехарактерно; наблюдается тромбоз в различных органах, появляется боль различной локализации (в почках, сердце, брюшной полости) за счет развития инфарктов; иктеричность склер и кожных покровов выражена слабо; уровень свободного гемоглобина в сыворотке крови резко повышен, а гаптоглобина, напротив, резко понижен; в моче определяется свободный гемоглобин, а спустя несколько суток — гемосидерин; выражены симптомы интоксикации (ознобы, лихорадка).
Таким образом, анализ клинических и лабораторных данных помогает разграничить внутриклеточный и внутрисосудистый гемолиз, приблизиться к определению варианта гемолитической анемии. Так, внутриклеточный гемолиз более свойствен мембранопатиям, а качественные гемоглобинопатии и приобретенные аутоиммунные гемолитические анемии протекают с внутрисосудистым гемолизом.
Гемолитические анемии занимают в структуре анемий 11,5 %, т.е. встречаются значительно реже, чем железодефицитные анемии. Некоторые формы гемолитических анемий распространены у людей определенных этнических групп. Однако, учитывая значительную миграцию населения, врач может встретиться с такой формой гемолитической анемии, которая не свойственна населению Украины.
Микросфероцитарная гемолитическая анемия.
Заболевание распространено повсеместно; ее частота в популяции составляет 1:5000. Наследственный микросфероцитоз передается по аутосомно-доминантному типу, реже — по аутосомно-рецессивному; в 25 % случаев наблюдаются спорадические случаи за счет новой мутации. Заболевание было впервые описано в 1871 г. Минковский (1900) и Шоффар (1907) выделили его в самостоятельную нозологическую форму и установили его наследственный характер.
Патогенез связан с дефектом спектрина, анкирина, белков 4,1 и 4,2, — с их дефицитом или отсутствием. Это приводит к тому, что мембрана эритроцита приобретает вид сеточки, через отверстия которой свободно входят и выходят многие активные вещества, необходимые для обеспечения стабильности мембраны. При этом нарушается электролитный обмен, поскольку в эритроцит в повышенном количестве проникают натрий и вода, вследствие чего эритроцит набухает, становится большим, приобретает шаровидную форму. В последующем размеры эритроцита уменьшаются при прохождении (пассаже) через синусоиды селезенки, его мембрана «обтесывается» с поверхности, а эритроцит уменьшается в размерах (микроцитоз), сохраняя сферическую форму.
Клиника. При объективном исследовании выявляют деформацию черепа, полидактилию, высокое, «готическое», нёбо. Эти изменения обусловлены расширением плацдарма кроветворения, который перемещается в период роста с плоских костей на трубчатые. Кожные покровы и видимые слизистые в разной степени желтушны, что зависит от фазы болезни: гемолитический криз или ремиссия. Увеличена селезенка, иногда и печень; нередки холелитиаз и приступы желчной колики.
Картина крови. Анемия нормохромная. Концентрация гемоглобина вне гемолитического криза сохраняется на уровне 90-100 г/л, а во время криза снижается до 40-50 г/л. Эритроциты малых размеров, имеют шаровидную форму, не определяется центральное просветление (микросфероцитоз). Количество ретикулоци-тов увеличено как в период ремиссии, так и (особенно) после гемолитического криза — 10-15 и 50-60% соответственно. Количество тромбоцитов остается в норме; количество лейкоцитов увеличивается в период криза, иногда наблюдается ядерный сдвиг до юных форм; СОЭ увеличена за счет уменьшения количества эритроцитной массы. Осмотическая резистентность (стойкость) эритроцитов понижена: их гемолиз начинается уже в 0,78% растворе натрия хлорида. В сомнительных случаях рекомендуется предварительная инкубация эритроцитов в течение суток, после чего их хрупкость повышается. Можно изучать спонтанный лизис эритроцитов после двухсуточной инкубации в стерильных условиях: если в норме разрушается от 0,4 до 5 % эритроцитов, то при микросфероцитарной анемии — 30-40%. Если к эритроцитам добавить глюкозу, то их аутолиз у здорового человека снижается до 0,03-0,4%, а у больных микросфероцитарной гемолитической анемии — до 10 %. В то же время микросфероциты более устойчивы в кислой среде, чем нормальные эритроциты.
В биохимическом анализе крови чаще всего увеличено содержание свободного билирубина, но не всегда. Так, если функциональная способность печени сохранена, а гемолиз невелик, то за счет связывания билирубина с глюкуроновой кислотой обеспечивается нормальный уровень свободного и связанного билирубина. Концентрация свободного билирубина закономерно повышается После гемолитического криза, который может развиться после случайной инфекции; в этих условиях наблюдается массовый распад эритроцитов и печень «не Успевает» связать свободный билирубин с глюкуроновой кислотой с образованием связанного билирубина. К гемолизу может присоединиться механическая желтуха, обусловленная образованием пигментных желчных камней в желчных протоках; в этих случаях увеличивается содержание обеих фракций билирубина; в моче повышается содержание уробилиногена, а в кале — стеркобилина. Гемолитический криз сопровождается активацией эритропоэза: в пунктате костного мозга выраженная нормобластическая реакция. Описывают отдельные случаи апластического гемолитического криза, когда отсутствует ответная активация эритропоэза, в костном мозге количество клеток эритроидного ростка уменьшено. Чаще такое состояние наблюдается на фоне развившейся инфекции.
Течение микросфероцитоза у гомозиготов, как правило, тяжелое, проявляется с детства, а у гетерозиготов протекает субклинически и возникает поздно, иногда после 20-30 лет. Описаны и более редкие формы мембранопатий.
Наследственный эллиптоцитоз наследуется аутосомно-доминантным путем; его частота варьирует от 0,02 до 0,05 % в популяции среди различных этносов мира. При электрофорезе у части больных отсутствует белок полосы 4,1. Эритроциты имеют форму эллипса, их деформируемость снижена, в связи с чем они быстро разрушаются в селезенке.
Течение в подавляющем большинстве случаев (95%) бессимптомное. Однако следует помнить, что наличие эллиптоцитоза не всегда свидетельствует о его наследственном характере, поскольку у здорового человека около 15% эритроцитов также имеют эллипсовидную форму. Клинически в манифестных случаях наблюдаются желтушность кожи и склер, спленомегалия, нередко диагностируются холелитиаз, изменения костного скелета.
Лабораторная диагностика основана на выявлении эллиптоцитов, которые иногда имеют форму палочки. Если в норме соотношение взаимно перпендикулярных диаметров эритроцита приближается к 1, то при эллиптоцитозе оно снижается до 0,78. Могут встречаться мишеневидные эритроциты, а эллиптоциты могут иметь различные размеры и нормохромную окраску. Цветовой показатель не отклоняется от нормы, уровень гемоглобина даже у гомозиготов не бывает низким, варьируя от 90 до 120 г/л. Количество ретикулоцитов увеличивается умеренно — до 4 %; осмотическая резистентность эритроцитов (ОРЭ) чаще понижена, но может быть и нормальной; в последнем случае проводят пробы с инкубацией эритроцитов и пробу на аутолиз, которые обнаруживают снижение ОРЭ.
Наследственный стоматоцитоз встречается среди людей всех этнических групп с невыясненной частотой, наследуется аутосомно-доминантным путем. Патогенез гемолиза при стоматоцитозе обусловлен несбалансированностью коэффициента калий/натрий в эритроците: калия накапливается меньше, чем натрия; возникающая при этом гипергидратация эритроцита снижает содержание в нем гемоглобина, а при окраске в центре эритроцита формируется просветление, напоминающее очертания рта. В некоторых случаях дисбаланс между калием и натрием в эритроците меняется и вместо гипергидратации возникает дегидратация, происходит «сгущение» гемоглобина в клетке, а при окраске эритроцит приобретает мишеневидную форму. Если эти клетки поместить в гипотонический раствор, то они принимают фору стоматоцита. ОРЭ, как правило, снижена; эритроциты разрушаются в селезенке, особенно у больных с резус-отрицательной кровью. Клиника в манифестных случаях аналогична другим наследственным гемолитическим анемиям. Выраженность анемии и желтухи умеренная, спленомегалия развивается только при длительном гемолизе. Концентрация свободного билирубина умеренно повышена, уровень гемоглобина обычно не падает ниже 90 г/л.
К более редким формам мембранопатий относятся наследственный акантоцитоз и пиропикноцитоз. Их клиника в выраженных случаях аналогична таковой при других наследственных гемолитических анемий. Основным диагностическим тестом при пиропикноцитозе является морфологическое исследование эритроцитов, которые выглядят искривленными и сморщенными, а в пробе с пиротестом (нагревание до 49-50 °С) их гемолиз наступает уже при температуре, которая на 3-4 °С ниже (эритроциты здорового человека разрушаются только при температуре 49-50 °С).
Акантоциты получили свое название благодаря наличию у них многочисленных выростов по всей поверхности, что обусловлено диспропорцией в содержании различных липидов: в клеточной мембране у них преобладает ригидный лецитин над более текучим сфингомиелином. Появление акантоцитов — это типичный признак акантоцитоза, но его нельзя считать патогномоничным для этой формы анемии, поскольку они могут встречаться и при тяжелой патологии печени, алкоголизме, микседеме, некоторых неврологических заболеваниях. О роли наследственности в этих случаях будет свидетельствовать наличие гемолитической анемии с умеренно повышенным уровнем ретикулоцитов и свободного билирубина.
Ферментопатии — несфероцитарные гемолитические анемии обусловлены наследственным снижением активности эритроцитарных ферментов или их нестабильностью. Эти формы гемолитических анемий наследуются аутосомно-рецессивным путем или по Х-сцепленному рецессивному типу. При них не встречаются ни морфологические изменения эритроцитов, ни нарушения ОРЭ.
Ферментопатии, связанные с дефицитом активности Г-6-ФД, распространены у жителей средиземноморского побережья, у евреев-сефардов, а также в Африке и Латинской Америке и в бывших малярийных районах Средней Азии и Закавказья. Полагают, что в этих географических регионах произошел естественный отбор: люди с нормальным составом ферментов в мембранах эритроцитов погибали чаще от малярии, чем люди с дефектным содержанием ферментов, поскольку они оказались более устойчивы к малярийному плазмодию. Дефицит активности Г-6-ФД у русских в нашей стране встречается в 2 % случаев.
Патогенез. В условиях дефицита Г-6-ФД нарушается метаболизм глутатиона, снижается его содержание в мембране эритроцита и накапливается перекись водорода, под влиянием которой гемоглобин и белки мембраны денатурируются; в эритроцитах появляются тельца Гейнца-Эрлиха, содержащие денатурированный гемоглобин. Эритроциты разрушаются как в кровеносном русле, так и в клетках ретикулоэндотелиальной системы.
Клинически заболевание имеет хроническое течение в форме несфероцитарной гемолитической анемии, преимущественно у жителей Северной Европы, реже — в виде острого внутрисосудистого гемолиза, чаще всего после приема провоцирующего пре
парата с окислительными свойствами (противомалярийные средства, сульфаниламиды), а также на фоне инфекции. Симптоматика криза: лихорадка, увеличение печени, выделение мочи черного цвета, резко окрашенного кала. Селезенка остается в норме. Вариант фавизма характеризуется кризовым течением, развивающимся после употребления в пищу конских бобов или вдыхания их пыльцы. При этом больные жалуются на слабость, озноб, боль в пояснице; рвота появляется через несколько часов или дней после действия провоцирующих факторов.
Лабораторная диагностика: нормохромная, регенераторная анемия; анизопойкилоцитоз, нормоциты, осколки эритроцитов (шизоциты); в эритроцитах — тельца Гейнца-Эрлиха. В биохимическом анализе крови повышено содержание свободного билирубина, наблюдается гипогаптоглобулинемия. Костномозговой пунктат характеризуется выраженной нормобластической реакцией: до 50 — 70 % клеток пунктата составляют элементы красного ростка. Диагноз подтверждают после установления дефицита фермента Г-6-ФД в эритроците в период компенсации процесса у больного, а также у его родственников.
Дефицит активности пируваткиназы как причина гемолитической анемии встречается с частотой 1:20 000 в популяции во всех этнических группах; наследуется аутосомно-рецессивным путем, проявляется несфероцитарной гемолитической анемии. В ее патогенезе имеет значение блокада гликолиза с нарушением синтеза АТФ, которая приводит к дефекту клеточной мембраны эритроцита. Гемолиз происходит внутриклеточно.
Клиника: бледность и желтушность кожных покровов, спленомегалия. Встречаются как полностью компенсированные, так и тяжелые формы заболевания. В гемограмме: нормохромная анемия, анизо- и пойкилоцитоз, могут быть макроциты, овалоциты, акантоциты, пиропикноциты. Отсутствуют сфероцитоз эритроцитов и тельца Гейнца-Эрлиха. Диагноз устанавливают на основании сниженной активности пируваткиназы в эритроцитах больного и у его родственников.
К этой форме гемолитических анемий относятся наследственные аномалии синтеза гемоглобина, обусловленные изменением первичной структуры его молекулы (качественные гемоглобинопатии) или нарушением соотношения (или синтеза) одной из цепей глобина с неизмененной первичной его структурой (количественные гемоглобинопатии). Это многочисленная группа заболеваний: выявлено уже более 500 аномальных гемоглобинов (т.е. качественных гемоглобинопатии) и более 100 различных типов ß-талассемий, а также несколько типов а-талассемий (т.е. количественных гемоглобинопатии). По данным ВОЗ (1983), около 200 тыс. детей ежегодно рождаются и погибают от гемоглобинопатии различного вида, а 240 млн гетерозиготных носителей гемоглобинопатии, не будучи больными, в потомстве могут иметь тяжело больных детей. Распространение гемоглобинопатии, как и других наследственных гемолитических анемий, соответствует ареалу распространения малярии. Среда множества гемоглобинопатии есть часто встречающиеся: талассемия, серповидноклеточная гемолитическая анемия, гемоглобинопатии С, Е, D и редко встречающиеся — метгемоглобинемия, нестабильные гемоглобины и др.
Талассемия — это мишеневидноклеточная анемия с нарушенным соотношением НЬА и HbF по биохимическим показателям; при этом возможна частичная недостаточность определенной цепи или ее полное отсутствие при преобладании другой цепи. Так, при нарушении синтеза ß-цепи будут преобладать а-цепи и наоборот. Бета-талассемия обусловлена снижением продукции ß-цепей гемоглобина. Неповрежденные а-цепи избыточно накапливаются в клетках эритропоэза, что ведет к повреждению мембраны и разрушению как клеток эритроидного ряда в костном мозге, так и эритроцитов в периферической крови; развиваются неэффективный эритропоэз и гемолиз с гипохромией эритроцитов, ибо содержание гемоглобина в эритроцитах недостаточно. Первыми описали ß-талассемию американские педиатры Кули и Ли в 1925 г. Тяжелая гомозиготная форма ß-талассемии получила название болезни Кули, или большой талассемии. Кроме того, по выраженности анемии и других клинических симптомов выделяют промежуточную, малую и минимальную талассемию. Помимо стран Средиземноморья талассемия встречается во Франции, Югославии, Швейцарии, Англии, Польше, а также у жителей Закавказья и Средней Азии, где в некоторых регионах частота носительства достигает 10-27 %.
Патогенез ß-талассемии связан с мутацией в локусе ß-глобина на 11-й паре хромосом, нарушающей синтез ß-глобиновой цепи. Вследствие неадекватного синтеза гемоглобина развивается гипохромная анемия. Преципитаты избыточного количества а-цепей удаляются из эритроцитов и эритрокариоцитов клетками ретикулогистиоцитарной системы; при этом клетки повреждаются и быстрее разрушаются. Таков механизм неэффективного эритропоэза и гемолиза эритроцитов и ретикулоцитов; гибель последних происходит в селезенке. При ß-талассемии накапливается также HbF, обладающий большим сродством к кислороду; однако отдача его тканям затруднена, что приводит к их гипоксии. Неэффективный эритропоэз способствует расширению плацдарма кроветворения, что отражается на структуре скелета; вместе с тем деструкция эритрокариоцитов в костном мозге ведет к повышенному всасыванию железа и патологической перегрузке организма железом. Гематологические признаки ß-талассемии иногда выявляются у больных анемией среди русских.
Клиника большой талассемии проявляется уже в детстве. У больных детей своеобразный башенный череп, монголоидное лицо с увеличенной верхней челюстью. Ранний признак болезни Кули — сплено- и гепатомегалия, развивающиеся за счет экстрамедуллярного кроветворения и гемосидероза. Со временем у них формируются цирроз печени, сахарный диабет в результате фиброза поджелудочной железы, а гемосидероз миокарда приводит к застойной сердечной недостаточности.
При анализе крови определяется гипохромная гиперрегенераторная анемия разной степени тяжести. В мазке крови обнаруживают гипохромные эритроциты малых размеров, мишеневидные, различной формы; много нормоцитов. В биохимическом анализе крови выявляются гипербилирубинемия за счет свободной фракции, гиперсидеремия, снижение ОЖСС, повышение активности ЛДГ. В эритроцитах повышен уровень фетального гемоглобина.
Альфа-талассемия распространена преимущественно в Юго-Восточной Азии, Китае, Африке и в Средиземноморье. Синтез а-цепей кодируют 4 гена, поэтому степень нарушения их синтеза меньше, чем при ß-талассемии; выраженный дисбаланс развивается только тогда, когда поражены все 4 гена. В то же время агрегаты из β-цепей, количество которых при α-талассемии обнаруживают в избытке, более растворимы, чем агрегаты из α-цепей, поэтому гемолиз при α-талассемии выражен слабее, чем при ß-талассемии, а эритропоэз более эффективен. Следовательно, клинические и лабораторные данные при α-талассемии выражены менее отчетливо, чем при ß-талассемии; их основное отличие в биохимическом составе гемоглобина эритроцитов: при α-талассемии уменьшено содержание α-цепей гемоглобина.
Качественные гемоглобинопатии представлены прежде всего серповидно-клеточной анемией, реже — анемией, обусловленной наличием нестабильных гемоглобинов; гомозиготной анемией СС, ЕЕ и др.; бессимптомной гемоглобинопатией НЬС и др., а также обусловленной присутствием гемоглобина М-группы. Более полно изучена серповидноклеточная анемия, широко распространенная у жителей тропической Африки, реже она встречается у жителей Средиземноморья, Ближнего и Среднего Востока, в Южной Америке. Важно отметить, что она распространена преимущественно в регионах, неблагополучных по малярии.
Качественные изменения гемоглобина состоят в том, что в положении ß-цепи глутаминовая кислота заменена на валин. Патологический гемоглобин получил название от слова «sicsle» — серп, потому что эритроцит приобретает серповидную форму. HbS менее растворим, чем нормальный, и в условиях гипоксии выпадает в осадок. В части случаев наблюдается бессимптомное носительство HbS, когда его концентрация в эритроцитах составляет не более 20-45 %; в обычных условиях гемолиз не развивается, спровоцировать его могут лишь особые ситуации, например полет на самолетах или подводное плавание, когда возникает гипоксия. Патогенез гемолиза сложен. Полагают, что в условиях кислой среды в молекуле HbS происходит смена отрицательного заряда на нейтральный, поскольку валин имеет нейтральную реакцию, а глутаминовая кислота — кислую. При этом растворимость гемоглобина снижается, образуются гидрофобные связи между остатками валина, а молекулы патологического HbS кристаллизуются, что приводит к образованию серповидной формы эритроцита.
Часть деформированных эритроцитов изменяется необратимо и преждевременно разрушается фагоцитами. Циркулируя в крови, серповидные эритроциты склеиваются между собой, особенно в мелких сосудах, образуя тромбы и нарушая кровообращение. Возникшая в этих условиях гипоксия способствует образованию новых серповидных эритроцитов — развивается своего рода порочный круг, результатом которого являются стаз и тромбоз в различных органах, в т.ч. в селезенке, которая постепенно атрофируется после гипертрофии. Генетический дефект локализуется в структурном гене ß-цепи, где произошла точечная мутация.
Клиника. Серповидноклеточная анемия клинически характеризуется симптомами, вызванными, с одной стороны, тромбозом сосудов различных органов серповидными эритроцитами, а с другой — гемолитической анемией. Степень тяжести анемии зависит от концентрации HbS в эритроците: чем она больше, тем ярче и тяжелее симптоматика. Кроме того, в эритроцитах могут присутствовать и другие патологические гемоглобины: HbF, HbD, НЬС и др. Иногда серповидноклеточная анемия сочетается с талассемией, при этом клинические проявления могут уменьшаться или, напротив, нарастать.
В первоначальном периоде болезни поражается преимущественно костномозговая система: появляется припухлость, а также боль за счет тромбоза сосудов, питающих сустав и кость. Возможен асептический некроз головки бедренной кости с присоединением в дальнейшем инфекции и остеомиелита. Гемолитические кризы развиваются обычно после перенесенных инфекций, имеют регенераторный либо гипорегенераторный характер и являются основной причиной смерти этих больных. В редких случаях наблюдается секвестрационный криз за счет депонирования крови в селезенке и печени, который выражается болевым абдоминальным синдромом за счет быстрого увеличения этих органов и сопровождается коллапсом; при этом гемолиз может отсутствовать, встречается легочный инфаркты в связи с нарушением микроциркуляции на уровне легочных сосудов.
Во втором периоде постоянный симптом — гемолитической анемии. Развивающаяся в трубчатых костях гиперплазия костного мозга (в них совершается активное кроветворение как компенсаторная реакция на гемолиз) сопровождается характерными изменениями скелета: тонкие конечности, искривленный позвоночник, башенный череп с выпуклостями в области лба и теменной кости. Гепато- и спленомегалия развиваются за счет активации в них эритропоэза, а также вторичного гемохроматоза и тромбоза; у части больных формируется желчнокаменная болезнь. Гемосидероз сердечной мышцы приводит к сердечной недостаточности, а гемосидероз печени, поджелудочной железы — к циррозу печени и сахарному диабету.
Тромбоз сосудов почек протекает с гематурией и последующей почечной недостаточностью. Неврологическая симптоматика обусловлена инсультом, параличом черепных нервов и др. Характерны трофические язвы на нижних конечностях. Большинство больных с тяжелой формой серповидноклеточной анемии погибает в течение 5 лет, а пережившие этот срок вступают в третий период, который характеризуется признаками нерезко выраженной гемолитической анемии. Селезенка у них обычно не прощупывается, так как повторные инфаркты приводят к ее сморщиванию — аутоспленэктомии. Печень остается увеличенной, неравномерно уплотненной, а частые инфекции принимают нередко септическое течение.
Гематологические изменения. Концентрация гемоглобина снижается ( 9 /л. В костном мозге наблюдается гиперплазия эритроидного ростка. Для выявления серповидности эритроцитов проводят специальную пробу: каплю крови покрывают стеклом, герметизируют, для чего края стекла смазывают вазелином; через несколько минут парциальное давление кислорода в капле крови под стеклом снижается и эритроциты принимают серповидную форму. Более информативен электрофорез гемоглобина: при серповидноклеточной анемии у гомозигот основную массу составляет HbS, HbA отсутствует, содержание HbF повышено; у гетерозигот при электрофорезе наряду с HbS выявляют НЬА. В крови повышено содержание свободной фракции билирубина, увеличено содержание сывороточного железа; ОРЭ повышена.
Гетерозиготные больные чувствуют себя практически здоровыми; анемию и морфологические изменения эритроцитов обнаруживают у них только в условиях гипоксии (подъем в горы, тяжелая физическая нагрузка, полет на самолетах и т.п.). Однако гемолитический криз и у них может закончиться летально.
Таким образом, клиника серповидноклеточной анемии характеризуется полисимптомностью: желтушностью кожных покровов, гипоксическим синдромом, гепатоспленомегалией, деформацией скелета, повторным тромбозом органов; из гематологических симптомов: анемией регенераторного характера, серповидностью эритроцитов, выявляемой при специальных пробах, гипербилирубинемией за счет свободной фракции. Принадлежность человека к определенной этнической группе дает основание заподозрить это заболевание и начать целенаправленное обследование для подтверждения или исключения этой анемии.
Приобретенные гемолитические анемии разделяют на анемии, связанные с воздействием антител, анемии, связанные с соматической мутацией, анемии, связанные с механическим или химическим повреждением эритроцитов, а также обусловленные разрушением эритроцитов паразитами или дефицитом витаминов.
Иммунные гемолитические анемии включают 4 варианта: изоиммунные, трансиммунные, гетероиммунные и аутоиммунные. Изоимунный вариант наблюдается в тех случаях, когда реципиенту перелиты эритроциты и клетки донора, несовместимые по системе АВО. При этом клетки крови донора разрушаются антителами, имеющимися у реципиента и направленными против антигена донора, т.е. гемолизу подвергаются эритроциты донора в крови реципиента (больного). Кроме того, подобная ситуация возможна при антигенной несовместимости клеток крови матери и плода. Трансиммунная гемолитическая анемия развивается у плода, имеющего одинаковые антигены с материнскими, если мать страдает аутоиммунной гемолитической анемией; при этом антитела матери через плаценту проникают в организм плода и оказывают разрушительное действие на его эритроциты. Гетероиммунная гемолитическая анемия отличается от предыдущей тем, что антитела направлены не против эритроцитов, а против антигена, фиксированного на эритроцитах; в ходе реакции антиген-антитело разрушаются и эритроциты. Антитела, к примеру, могут быть направлены против лекарственного препарата, фиксированного на эритроцитах. Гетероиммунной считается также гемолитическая анемия при наличии эритроцитов с измененной антигенной структурой; в этих случаях эритроцит становится «чужим» для организма и иммунная система целенаправленно организует специфическую защиту (синтезируются антитела против измененной антигенной структуры эритроцита).
Об аутоиммунном характере гемолитической анемии свидетельствуют лишь те случаи, когда образующиеся антитела направлены против собственных неизмененных антигенов, конкретно — против нормальной антигенной структуры клеток эритропоэза: эритрокариоцитов или эритроцитов периферической крови. Антитела могут быть представлены неполными тепловыми агглютининами, тепловыми гемолизинами, полными холодовыми агглютининами и двухфазными гемолизинами.
Патогенез аутоиммунной гемолитической анемии. Сущность аутоиммунных процессов состоит в том, что в результате ослабления Т-супрессорной системы иммунитета, контролирующей аутоагрессию, происходит активация В-системы иммунитета, синтезирующей при этом антитела против неизмененных антигенов различных органов. В реализации аутоагрессии принимают участие также Т-лимфоциты-киллеры. Антитела — это иммуноглобулины (Ig), принадлежащие чаще всего к классу G, реже — М и А; они специфичны и направлены против определенного антигена. К IgM относятся, в частности, холодовые антитела и двухфазные гемолизины. Эритроцит, несущий на себе антитела, фагоцитируется макрофагами и в них разрушается; возможен лизис эритроцитов с участием комплемента. Антитела класса IgM могут вызвать агглютинацию эритроцитов непосредственно в кровеносном русле, а антитела класса IgG способны разрушать эритроцит только в макрофагах селезенки. Во всех случаях гемолиз эритроцитов происходит тем интенсивнее, чем больше на их поверхности находится антител. Описана гемолитическая анемия с антителами к спектрину.
Клиника аутоиммунной гемолитической анемии различается в зависимости от вида антител. Чаще других встречается аутоиммунная гемолитическая анемия с неполными тепловыми агглютининами. Заболевание может иметь острое, внезапное начало или сразу же принять хроническое течение. В острых случаях повышается температура тела; кожные покровы более бледны, чем желтушны; могут быть умеренно увеличены периферические лимфатические узлы, селезенка и печень. Анемический синдром сопровождается тахикардией, одышкой, общей слабостью. В общем анализе крови выраженность анемии может быть различной; характер ее обычно нормохромный, умеренно регенераторный; в мазке крови нередко появляются нормобласты, а в период криза наблюдается лейкоцитоз (реже — лейкопения), нередко — тромбоцитопения. В биохимическом анализе крови: умеренная гипербилирубинемия за счет свободной фракции; ОРЭ умеренно снижена. Решающее диагностическое значение имеет положительная прямая проба Кумбса, выявляющая антитела, фиксированные на эритроцитах. При этом следует иметь в виду, что при небольшом количестве антител на эритроцитах, а также после гемолитического криза, когда количество эритроцитов снижается, или после приема глюкокортикоидных гормонов проба Кумбса может быть отрицательной. В этих случаях проводят модифицированную пробу, суть которой состоит в том, что эритроциты 0(1) резус-положительной группы крови наслаивают на агрегированные белки иммунной сыворотки, добавляя их к отмытым эритроцитам больного, — при наличии антител эритроциты склеиваются.
Пароксизмальная холодовая гемоглобинурия и аутоиммунная гемолитическая анемия с полными холодовыми агглютининами характеризуются тем, что у больных антиэритроцитарные антитела проявляют свои агглютинирующие или гемолизирующие свойства при охлаждении. В первую фазу наблюдается фиксация антител на эритроцитах при температуре 0-15 °С; во вторую — наступает внутрисосудистый гемолиз при температуре 37 °С. Холодовые агглютинины в низких титрах присутствуют у 95 % здоровых людей. Холодовые антитела принадлежат к классу IgM; они активируют комплемент крови в дистальных отделах конечностей, так как в них, как правило, температура ниже, чем на других участках тела, — развивается синдром Рейно. Гемолитический криз чаще всего связан с инфекцией микоплазмой (известно, например, что микоплазменная пневмония может осложняться аутоиммунной гемолитической анемией), вирусом Эпштейна-Барр. Клиника соответствует симптоматике внутрисосудистого гемолиза. После гемолитического криза может развиться хроническая форма гемолитической анемии. Первично-хроническая гемолитическая анемия с холодовыми агглютининами развивается иногда у пожилых людей с хроническим лимфолейкозом или парапротеинемическими лейкозами; она носит внутрисосудистый характер. Эритроциты, нагруженные Холодовыми антителами, разрушаются в печени.
Пароксизмальная холодовая гемоглобинурия (синдром Доната-Ландштейнера) характеризуется присутствием антител класса IgG, которые активны в холодной среде и при температуре 37 °С способны активизировать комплемент. Этот вариант гемолитической анемии связан с поздней стадией сифилиса и другими инфекциями, но может быть спровоцирован и локальным охлаждением, например приемом холодной воды. Клиника характерна для внутрисосудистого гемолиза; патогномоничный синдром — боль в поясничной области и моча цвета мясных помоев или темно-коричневой окраски. В моче находят гемоглобин и гемосидерин. Анемия нормохромная, регенераторная; по лабораторным и клиническим данным имеются признаки ДВС-синдрома и почечной недостаточности. В крови увеличено содержание свободной фракции билирубина.
Пароксизмальная ночная гемоглобинурия Маркиафавы—Микели обусловлена приобретенным дефектом мембраны эритроцитов, которая становится чувствительной к нормальному комплементу крови в условиях ацидоза. Дефект мембраны эритроцитов является результатом соматической мутации на ранних этапах клеточной дифференцировки, что подтверждается одновременными тромбоцито- и лейкопенией вследствие неполноценности их мембраны. Клиника пароксизмальной ночной гемоглобинурии характеризуется кризами: они появляются после сна (ночью имеет место физиологический ацидоз), протекают с выраженным болевым синдромом в поясничной области, выделением мочи черного цвета, в которой при исследовании обнаруживают гемосидерин, свободный гемоглобин и железо. Возможны тромботические осложнения: тромбоз периферических вен, воротной вены, вен печени (синдром Бадда-Киари), а также брыжеечных и церебральных вен. Пароксизмальная ночная гемоглобинурия может быть ассоциирована с лейкозами и апластической анемией. Кризы иногда провоцируются инфекцией, приемом препаратов железа. Лабораторные признаки: нормо- или гипохромная анемия, умеренная лейко- и тромбоцитопения. В крови повышен уровень свободного билирубина, содержание сывороточного железа понижено (за счет потери его с мочой, чем обусловлена и гипохромия эритроцитов) или остается в норме. Пробы на кислотную устойчивость эритроцитов (тест Хема) и гемолиз с добавлением глюкозы (сахарозный тест Гармана) выявляют пониженную устойчивость эритроцитов, что патогномонично для пароксизмальной ночной гемоглобинурии.
Гемолитическая анемия, обусловленная механическими факторами, связана с разрушением эритроцитов при их прохождении через измененные сосуды или через искусственные клапаны. Эндотелий сосудов изменяется при васкулитах, злокачественной артериальной гипертензии; при этом адгезия и агрегация тромбоцитов активированы, как и система свертывания крови и образования тромбина. Развиваются распространенный стаз крови и тромбоз мелких кровеносных сосудов (ДВС-синдром) с травматизацией эритроцитов, в результате чего они фрагментируются; в мазке крови находят многочисленные фрагменты эритроцитов (шистоциты). Разрушаются эритроциты также при их прохождении через искусственные клапаны (чаще — при многоклапанной коррекции); описана гемолитическая анемия на фоне сенильного кальцинированного аортального клапана. Диагноз базируется на признаках анемии, повышении концентрации свободного билирубина в сыворотке крови, наличии шистоцитов в мазке периферической крови и симптоматике основного заболевания, ставшего причиной механического гемолиза.
План обследования больного при подозрении на гемолитическую анемию.
План обследования больного можно условно разделить на два этапа:
общие для всех гемолитических анемий лабораторно-инструментальные исследования
Первый этап:
Общий анализ крови с определением количества ретикулоцитов и морфологическим исследованием эритроцитов.
источник