Одним из самых редких, но тяжелых заболеваний системы кровообращения считается апластическая анемия, симптомы, причины, методы лечения этого вида малокровия бывают различны. Все зависит от формы, вида и степени тяжести данной патологии.

Люди, далекие от медицины, зачастую не имеют представления о том, что такое апластическая анемия. Так называется расстройство кроветворной функции костного мозга, ведущее к снижению выработки кровяных клеток. Это приводит к серьезным патологическим изменениям во всех системах человеческого организма. Очень часто заболевание заканчивается летально.
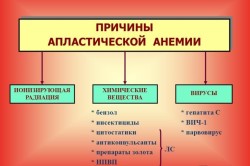
Этиология апластической анемии до сих пор досконально не изучена. Поэтому окончательный список причин данного заболевания все еще полностью не составлен. На данный момент основными причинами апластического малокровия считаются:
- плохая экология в регионе проживания;
- ионизирующая радиация;
- постоянные контакты с опасными химическими веществами в течение долгого времени;
- отдельные инфекционные заболевания: гепатит, цитомегаловирус;
- разрушение костного мозга под воздействием некоторых медикаментозных средств при их длительном приеме в высоких дозах, особенно левомицетина, мышьяка, бензола, химических препаратов, противоопухолевых средств, антибиотиков;
- генетическая предрасположенность;
- беременность;
- патологии костного мозга;
- злоупотребление алкоголем.
Возникновение апластической анемии у детей сразу после рождения может быть спровоцировано такими причинами, как:
- врожденный сифилис;
- токсоплазмоз;
- генерализованный мегалоцитоз.
Часто у детей диагностируется идиопатическая апластическая анемия. В этом случае определить точную причину возникновения патологии не представляется сегодня возможным.
При гипопластической анемии наблюдается еще более серьезное расстройство кроветворной функции костного мозга. В целом гипопластические и апластические анемии вызывают сходные патологии органов и систем организма, имеют похожую симптоматику и лечатся примерно одинаково.
Костный мозг образуется губчатой структурой и располагается в глубине трубчатых костей. Посредством его структуры происходит выработка различных клеток крови: лейкоцитов, тромбоцитов, эритроцитов.
 При помощи указанных клеток организуется защита организма от проникающих извне микроорганизмов и вызываемых ими заболеваний.
При помощи указанных клеток организуется защита организма от проникающих извне микроорганизмов и вызываемых ими заболеваний.
При апластической анемии, прежде всего, нарушается эритрогенез. Стимулировать этот процесс медикаментозными средствами невозможно. В этом случае может помочь только пересадка донорского костного мозга.
Помимо этого, апластичная анемия развивается на фоне уменьшения количества и изменения функций стволовых клеток.
Причиной этого становится нарушение микроокружения, клеточное или гуморальное воздействие иммунного характера, препятствующее нормальному функционированию стволовых клеток.
Апластическая анемия, симптомы которой часто незаметны, является серьезной патологией. В отдельных случаях недуг начинается в острой форме с быстрым нарастанием симптоматических проявлений. Развитие болезни может быстро прогрессировать, приводя к летальному исходу. Возможно также хроническое с периодическими ремиссиями и рецидивами протекание анемии.
Апластическая анемия симптомы имеет следующие: анемический, геморрагический и лейкоцитопенический синдромы. Для каждого из них характерны определенные признаки.
Анемический синдром (снижение уровня гемоглобина) возникает при нарушении формирования эритроцитов и сопровождается такими симптомами, как:
- общая слабость;
- сонливость;
- бессонница;
- раздражительность;
- одышка;
- шум в ушах;
- тахикардия;
- бледность кожных покровов;
- быстрая утомляемость.
Геморрагический синдром (повышение кровоточивости) вызван понижением уровня тромбоцитов в крови. Его сопровождают такие симптомы, как:
- петехии, т. е. точечные кровоизлияния на коже и слизистых оболочках и гематомы;
- плохая свертываемость крови;
- десневые и носовые кровотечения;
- появление крови в моче;
- обильные менструальные выделения у женщин.
 Недостаток лейкоцитов в крови вызывает развитие лейкоцитопенического синдрома.
Недостаток лейкоцитов в крови вызывает развитие лейкоцитопенического синдрома.
Он сопровождается следующими симптомами:
- появление хронических очагов инфекции в различных органах и на поверхности кожи больного;
- учащение инфекционных заболеваний, протекающих длительно и со многими осложнениями;
- стоматит;
- внезапная гипертермия без видимых причин.
По форме апластическая и гипопластическая анемия может быть как врожденной, так и приобретенной.
После проведения анамнестической беседы врач проводит общий осмотр пациента. При этом обязательно применяются такие методы, как:
- выслушивание (аускультация) сердечных шумов;
- простукивание (перкуссия) живота, показывающая увеличение таких органов, как печень и селезенка;
- прощупывание (пальпация) для уточнения результатов перкуссии.
Для получения дополнительных сведений больному назначается:
- общий анализ крови;
- биохимический анализ крови;
- клинический анализ крови — гемограмма;
- забор мазка — биопсия, костного мозга;
- анализ мазка костного мозга, на основании которого составляется миелограмма.
При необходимости больному проводятся специальное тестирование, помогающее уточнить причину заболевания.
На основании всех полученных данных составляется точная клиническая картина заболевания, необходимая для определения курса лечения.
Когда больному диагностируется апластическая анемия, лечение проводится по 3 направлениям: этиологическому, симптоматическому и патогенетическому. Если известна точная причина возникновения заболевания, ее необходимо ликвидировать: прекратить контакт с токсинами, сменить лекарственные препараты, используемые для лечения других заболеваний и т. д.
Восстановлению кроветворного функционирования костного мозга помогают следующие препараты:
- кортикостероиды;
- андрогены;
- цитоксины;
- иммуносупрессоры.
Помимо медикаментозной терапии лечение апластической анемии проводится при помощи таких процедур, как переливание крови и ТКМ. Посредством переливания донорской крови организм больного частично обеспечивается недостающими кровяными клетками.
В особых случаях больным проводится трансплантация костного мозга. Костный мозг донора должен максимально соответствовать костному мозгу реципиента. Поэтому этот метод особенно эффективен, если у больного есть однояйцевый близнец. Процент выживаемости после трансплантации костного мозга выше у реципиентов более молодого возраста.
 Степень полного выздоровления среди больных апластической анемией, к сожалению, невысока. В основном речь идет о продлении жизни больного и облегчении его страданий, вызванных болезнью.
Степень полного выздоровления среди больных апластической анемией, к сожалению, невысока. В основном речь идет о продлении жизни больного и облегчении его страданий, вызванных болезнью.
Основными факторами, влияющими на получение положительных эффектов, являются:
- Степень тяжести заболевания.
- Правильный выбор и хорошая переносимость лечения.
- Возраст больного. Легкая апластическая анемия у детей и молодых людей, не достигших 30-летнего возраста, полностью вылечивается чаще, чем у людей среднего и старшего возраста. При тяжелых формах болезни прогнозы не столь благоприятные.
Основным условием ремиссии и значительного продления жизненного срока является хорошая переносимость патогенетической терапии и успешное проведение трансплантации костного мозга.
Профилактика анемии различного вида может быть первичной или вторичной. Первичная профилактика предполагает исключение возможности развития данной патологии у относительно здорового человека. Этого можно добиться при помощи таких способов, как:
- ведение здорового образа жизни;
- правильное питание;
- постоянное укрепление иммунной системы;
- исключение или сведение к минимуму контакта с ионизирующими излучателями и токсичными веществами, вызывающими патологии костного мозга;
- контролированный прием лекарственных препаратов во время лечения других заболеваний;
- периодические визиты к терапевту и гематологу;
- полный отказ от употребления алкоголя и никотина.
Вторичная профилактика анемии проводится для замедления развития заболевания, избегания его осложнения сопутствующими патологиями. Для этого необходимо:
- аккуратное проведение лечения;
- постоянный контроль специалистов — гематолога и терапевта;
- тщательное контролирование больным своего состояния, немедленное посещение врача при появлении новых симптомов;
- постоянный прием поддерживающих препаратов в течение долгого времени.
Апластическая анемия — опасная патология, которую лечат годами. Только под наблюдением специалиста можно добиться стойкой ремиссии патологии. Самолечение в этом случае неприемлемо и может привести к плачевным последствиям.
источник
Мегалобластическая анемия — это патологическое состояние, заключающееся в общем клеточном расстройстве, в основе которого лежит нарушение клеточного деления за счет заниженного синтеза нуклеиновых кислот, что, в большинстве случаев, является результатом недостаточности витамина В12 или фолиевой кислоты. В гематологическом плане это заболевание характеризуется мегалобластическим преобразованием и неэффективностью миелопоэза.
Мегалобластическая анемия коренным образом отличается от гипохромной анемии, при которой нарушение касается клеточного созревания (за счет неполноценности синтезирования гемоглобина), равно как и от апластической анемии, при которой нарушается дифференциация «клеток-штамм» в кровяные клетки.
Основным нарушением, обусловливающим мегалобластическую морфологию является недостаточное синтезирование ДНК (биохимическими механизмами, описанными далее). Результаты этого дефицита следующие:
а) Увеличивается межмитотическая фаза, во время которой продолжается синтез РНК и белков. Это объясняет утрату количественного равновесия между ДНК и РНК, что, в морфологическом плане проявляется изменением отношения ядро-цитоплазма в пользу последней.
б) Кровяные клетки не подвергаются нормальному числу делений (в основном это касается последних трех делений эритробластического ряда) (Weicker, Rohr), при этом ядро остается большим и выглядит как «молодое », в то время как в цитоплазме развивается характеристика зрелой клетки (асинхронизм созревания ядра и цитоплазмы).
Нарушение механизма деления ядра проявляется атипическими митозами (многополюсными, с тонкими хромосомами, под острым углом и неоднородно распределенными), тенденцией к почкованию ядра и чрезмерным сегментированием (в гранулоцитном и мегакариоцитном рядах), аспектами кариорексиса и ядерных остатков (в эритроидном ряде); при этом наличие полиплоидных клеток (4n, 8n или даже более, с единым или множественным ядром) отражают тяжелый дефицит.
в) Отмеченное мегалобластическое расстройство обусловливает усиленное внутрикостномозговое разрушение этих клеток, пропорционально степени дефицита (неэффективность кроветворения, отражающаяся в высоком уровне лактикодегидрогеназы и билирубина, и ростом «ранней фракции» последней). Показатель разрушения находящихся в кровообращении элементов также высок, а средняя продолжительность жизни эритроцитов равняется 1/2—1/3 нормы.
г) Последствием неполноценного кроветворения является анемия, которая (под стимулирующим влиянием эритропоэтина) способствует развитию эритробластической гиперплазии (при изменении отношения Э:Г, росте клеточной костаномозговой массы и гематопоэтической преобразовании жирного костного мозга), что усиливает дефицит фолиевой кислоты или витамина В12 (за счет увеличенного расхода) и замыкает порочный круг усугублением мегалобластоза и анемии.
В основе количественных и качественных изменений остальных двух костномозговых рядов находятся те же нарушения, которые поражают и эритробластный ряд. Неэффективный грапулоцитопоэз поддерживается не только морфологическими альтерационными изменениями, но также повышением уровня мурамидазы в сыворотке (Perillie).
Неэффективность мегакариопоэза, отмечаемая при мегалобластной анемии, которую подсказывал контраст между ростом общей массы мегакариоцитов костного мозга и периферической тромбоцитопзнией, была подтверждена работами Harker и Finch, установивших среднюю выработку, на ядерную единицу, шести тромбоцитов вместо сорока (норма). Рост разрушения гранулоцитов и тромбоцитов на периферии — возможно путем увеличенной секвестрации селезенкой — может способствовать, в различной степени, развивающейся при этом цитопении.
Подобные клеточные альтерационные изменения наблюдаются и в других клеточных системах, в основном у тех, характерной чертой которых составляет коэффициент быстрого деления, в том числе эпителий пищеварительного тракта (в отдельных случаях и клетки зародыша, плодных придатков). В этом следует искать объяснение некоторых клинических признаков или осложнений (глоссит, затруднения пищеварения, расстройство поглощения, перинатальные осложнения и пр.).
Общим диагностическим элементом мегалобластных анемий является определение мегалобластной морфологии всех кровяных клеток костного мозга и периферической крови.
Термином мегалобласт определяются предшественники эритроцитов. В принципе мегалобласт крупнее соответствующего нормального эритробласта с измененным отношением ядро-цитоплазма в пользу последней. Ядро промегалобласта (диаметром 20—30 u) объемистое, нередко расположено эксцентрично, хроматинная структура в виде жемчужин, содержит несколько крупных ядрышек синеватой окраски; цитоплазма синего цвета, наиболее ясная зона расположена вокруг ядра, причем может сохраниться и на последующих стадиях.
На стадии базофильного мегалобласта ядро уменьшается, отсутствуют ядрышка, хроматинная структура более плотная, однако не наблюдаются характерные для базофильного нормобласта скопления, базофильность цитоплазмы станитовся более резкой. На стадии полихроматофильного мегалобласта отражен наиболее ярко асинхронизм созревания между ядром (еще крупным, с едва начинающим организоваться в виде блоков хроматинном) и цитоплазмой (растянутой, полихроматофильной, нередко с наличием телец Жолли). Диаметр оксифильного мегалобласта колеблется от 10 до 18 u, его цитоплазма полностью оксифильная и хроматинная структура ядра еще выявима.
Мегалоцит характеризуется более крупными размерами (от 12 до 14 u), отсутствием ясной центральной зоны (большей толщиной) и часто чуть овальной формой.
Происходящие в гранулоцитном ряде мегалобластические изменения отражаются в увеличенном, в принципе, размере клетки и обильности цитоплазмы. Эти признаки проявляются более четко в гигантских метамиелоцитах, имеющих крупное ядро, весьма скудную хроматинную структуру, не соответствующей форме ядра, при этом цитоплазма продолжает оставаться базофильной. Наблюдается тенденция к гиперсегментированию в результате чего образуется форма гиперсегментированных гранулоцитов (6, 8, 10 и более сегментов), хроматинная структура которых, однако, менее плотная (в отдельных случаях сегментирование частичное, в виде надреза).
Изменения мегакариоцитов менее четкие, в некоторых случаях их размеры отклоняются от нормы, зернистость недостаточная. Хроматинная структура менее плотная, естественная тенденция к сегментированию ядра усиливается, появляются отделившиеся сегменты или сегменты в виде «виноградной грозди» или «взрывных» мегакариоцитов. Тромбоциты крупнее (макро- или даже мегатромбоциты) с лучше выраженной структурой, чем нормальная.
Что касается этиопатогенеза мегалобластической анемии отмечаем, что значительные достижения последних двух десятилетий в изучении этого заболевания привели к выделению следующих трех основных категорий, из них первые две включают случаи, обусловливаемые недостатком витамина В12 и фолиевой кислоты (с возможным их сочетанием при определенных обстоятельствах), а третья — более редкие случаи этого заболевания, независимые от первых двух недостатков, вызываемые известными или еще неизвестными причинами.
источник
Термином «анемия» обозначают патологические состояния, характеризующиеся уменьшением содержания гемоглобина (Гб) и/или количества эритроцитов (Эр) в единице объема крови.
Анемический синдром выявляется в любом возрасте и является одной из самых распространенных патологий. Если учитывать все анемии не только как нозологические формы, но и анемический синдром при различных заболеваниях, то масштабы проблемы столь широки, что ее иногда характеризуют, как «скрытую эпидемию» («Анемия — скрытая эпидемия», 2004). Анемия выявляется у 15-20% беременных, а по некоторым данным — у 40% будущих матерей.
В зависимости от уровня гемоглобина выделяют анемию тяжелой (уровень гемоглобина 75 г/л и ниже), умеренной или средней (гемоглобин 80-100 г/л) и легкой (100-110 г/л) степени тяжести.
- Этиологически они подразделяются на анемии, обусловленные внутри-эритроцитарными факторами — обычно врожденными (аномалии мембраны, ферментопатии, гемоглобинопатии), и анемии, обусловленные внеэри-троцитарными факторами — обычно приобретенными.
- В зависимости от размеров эритроцитов — микроцитарные анемии (средний объем эритроцитов МСУ 3 ), нормоцитарные (СДЭ = 7-8 мкм;МСУ = 80-100 мкм 3 ) и макроцитарные (МСУ более 95-100 мкмЗ) МСУ анемии.
- В зависимости от степени насыщения гемоглобином — гипохромные (с цветовым показателем — ЦП — менее 0,85 и средней концентрацией гемоглобина в эритроцитах — МСНС — ниже 30 г/дл), нормохромные (ЦП = 0,9-1,1; МСНС = 30-38 г/дл) и гиперхромные (ЦП выше 1,1; МСНС более 38 г/дл) анемии.
- В зависимости от сохранности и адекватности реакции костного мозга на снижение уровня гемоглобина и эритроцитов, определяемого по числу ретикулоцитов, анемии могут быть разделены на гипорегенераторные (при уровне ретикулоцитов менее 1-1,2% при наличии анемии), связанные с нарушением продукции эритроцитов в костном мозге, а также нормо-или гиперрегенераторные (уровень ретикулоцитов повышен умеренно или значительно — до 20-30% и более. Повышение числа ретикулоцитов указывает на то, что малокровие, скорее всего, обусловлено гемолизом (т. е. повышенным разрушением эритроцитов) или кровотечением.
С учетом ведущего механизма развития строятся патогенетические классификации, примером которых может быть следующий вариант группировки анемий по патогенетическому механизму (Воробьев П. А., 2001):
- Железодефицитные анемии.
- Анемии, связанные с нарушением синтеза тема: сидероахрестические, дефицит гемсинтетазы.
- Анемии, связанные с нарушением синтеза ДНК — мегалобластные анемии.
- Анемии, обусловленные нарушением транспорта железа — атрансферри-немия.
- Гемолитические анемии.
- Анемии, связанные с костномозговой недостаточностью.
- Анемии, связанные с нарушением регуляции эритропоэза (повышение уровня ингибиторов эритропоэза).
зависят от степени снижения кислород-насыщающей способности крови, степени изменения общего объема крови, проявлений основного заболевания, которое приводит к развитию анемии и способности сердечно-сосудистой и дыхательной систем компенсировать анемию.
Многообразные клинико-гематологические проявления анемий можно разделить на две основные группы: симптомы, возникновение которых связано с гипоксией (так называемые неспецифические симптомы) и симптомы, характерные только для определенной анемии.
К общеанемическим симптомам, составляющим общеанемический синдром, относят слабость, бледность кожи и слизистых, одышку, тахикардию, головокружение, головную боль, снижение умственной концентрации, сонливость. Практически для всех видов анемий характерны симптомы со стороны сердечно-сосудистой системы, которые проявляются наличием шума в сердце, обычно систолического характера, который выслушивается в области легочной артерии. При тяжелой анемии шумы могут определяться в области митрального и трехстворчато го клапанов. Эти шумы легко дифференцируются от шумов, возникающих при органических поражениях сердца. При анемиях часто наблюдается ритм галопа пресистолического и протодиастолического типов. Изменения ЭКГ проявляются в депрессии интервала 8Т с 17-образной деформацией 8Т сегмента, изменении продолжительности электрической систолы (интервал С>Т), нарушении предсердно-желудочковой проводимости. При тяжелых анемиях (уровень НЬ ниже 60 -70 г/л) может наблюдаться фибрилляция предсердий.
При диагностике анемий важно выяснить особенности начала заболевания. Так постепенное начало чаще связано с нарушением продукции эритроцитов, острое — чаще наблюдается при повышенном разрушении красных кровяных клеток. Следует отметить имевшиеся провоцирующие факторы (вирусные инфекции, химические и физические факторы и др.), что может свидетельствовать в пользу определенного вида анемий (аутоиммунных, фер-ментопатий и т. д.).
Для установления патогенеза анемии при оценке показателей «красной крови» обращают внимание на так называемые, эритроцитарные параметры (индексы), отражающие размеры эритроцитов и степень их насыщения гемоглобином, количество ретикулоцитов и морфологические характеристики эритроидных клеток, которые отмечает врач-лаборант при просмотре мазка крови.
Снижение МСУ характерно для микроцитарных — железодефицитной анемии (ЖДА), талассемии. Причиной макроцитарной анемии, характеризующейся повышением показателя МСУ, могут быть мегалобластные анемии или нарушения, не связанные с нарушенным синтезом ДНК. Так причиной макроцитоза могут стать хронические заболевания печени, хронические заболевания почек, курение, гипо- и гипертиреоидизм.
Гипохромия эритроцитов выявляется в случаях, когда снижение Гб выражено сильнее, чем уменьшение числа Эр. Чаще всего это происходит при нарушениях процессов синтеза гемоглобина (при железодефицитной анемии, талассемии, свинцовом отравлении) и сидеробластной анемии (нарушение утилизации запасов железа). Как нормохромные, обычно характеризуются гемолитические анемии и анемии, связанные с гипопластическим состоянием костного мозга в частности. Гиперхромия — повышенная насыщенность гемоглобином цитоплазмы клеток характерна для макро- и мегалоцитов.
Гипорегенераторные анемии со сниженным или нормальным уровнем ретикулоцитов наблюдаются при дефиците железа, анемии при хронических заболеваниях или миелодисплазии. Значительное повышение числа ретикулоцитов указывает на то, что малокровие, вероятее всего, обусловлено гемолизом или кровотечением.
Важную информацию можно получить при оценке морфологических особенностей эритроцитов. Наличие макро- и особенно мегалоцитоза эритроцитов типично для Вр и фолиево-дефицитной анемии. Сфероциты встречаются при аутоиммунном гемолизе или наследственном сфероцитозе, шизоциты — фраг-ментированные эритроциты, расщепленные фибриновыми нитями — при микроангиопатиях (тромботической тромбоцитопенической пурпуре или диссемини-рованном внутрисосудистом свертывании — ДВС). Мишеневидные («таргетные») клетки в небольшом количестве появляются в крови при ряде гемоглобинопатии, при патологии печени, но наиболее характерны для талассемии, при которой их процентное содержание может быть значительным. Появление базофиль-ной пунктации эритроцитов характерно для свинцовых отравлений, талассемии и других дизэритропоэтических анемиях.
Ядерные формы эритроцитов (нормобласты или эритрокариоциты) наблюдаются при эритробластической анемии, инфильтрации костного мозга, гемолизе, гипоксии.
Дальнейшие исследования проводятся для уточнения предполагаемого варианта анемии и включают в себя биохимические, иммунологические и другие виды анализов.
Существуют определенные группы больных, относящихся к группам риска по развитию того или иного вида малокровия, которых желательно обследовать регулярно в порядке скрининга с целью выявления предрасположенности к анемии или ранних стадий анемии и проведения соответствующих профилактических и лечебных мероприятий.
Железодефщитная анемия (ЖДА) — наиболее распространенная форма анемий. Социальная значимость данной патологии определяется частой встречаемостью ЖДС среди женщин детородного возраста и детей, неблагоприятным влиянием железодефщита нарост и развитие детей и подростков, снижением работоспособности и ухудшением качества жизни взрослых, зависимостью частоты заболеваемости от ряда социальных факторов (уровня жизни, образования, здравоохранения).
Основные причины развития дисбаланса обмена железа в организме, ведущего к железодефицитным состояниям:
- Потери крови, особенно меноррагии или кровотечения из желудочно-кишечного тракта (ЖКТ) при эзофагите, пептической язве, карциноме, колите, дивертикулите, геморрое.
- Неадекватное питание, приводящее к развитию ЖДА у детей и подростков, реже у взрослых.
- Глистные инвазии и связанные с ними ЖК-кровопотери.
- Мальабсорбция (например, при кишечных заболеваниях).
К группам повышенного риска развития железодефицита относятся:
- Дети: потребности в железе при быстром росте часто превышают его поступление.
- Девочки в подростковом возрасте.
- Женщины: некомпенсированные потери железа во время менструаций, беременности, родов, гиперполименоррее.
- Доноры без компенсации потерь железа.
- Пожилые люди вследствие хронических гастроинтестинальных заболеваний и питания, содержащего мало мясных продуктов. Определенную роль в развитии заболевания играет геликобактерная инфекция.
Клиническая картина заболевания складывается из неспецифических проявлений общеанемического синдрома и проявлений тканевого дефицита железа — так называемого сидеропенического синдрома. Как правило, отмечается сухость кожи, характерный алебастровый или зеленоватый оттенок кожных покровов, а также голубоватый оттенок склер («симптом голубых склер»), как отражение дистрофических изменений роговицы в условиях дефицита железа, повышенная ломкость ногтей и волос. Возможно появление поперечной ис-черченности ногтевой пластинки и специфические «ложкообразные» их изменения — койлонихии. У больных имеется выраженная общая слабость, которая может не соответствовать степени анемии, и мышечная слабость, обусловленная нарушением синтеза миоглобина. Может выявляться дисфагия, извращение вкуса и обоняния с пристрастием к необычным запахам, «заеды» в углах рта (ангулярный стоматит), сглаженность сосочков языка, дизурические явления, недержание мочи при смехе, кашле.
Железодефицитная анемия сопровождается многочисленными осложнениями в течение беременности и родов как у матери, так и у плода, включая невынашивание беременности, кровотечение в родах.
Поскольку заболевание развивается медленно (месяцы и даже годы), клинические проявления обычно сглажены и больные адаптированы ко многим проявлениям.
Для анализов крови при ЖДА характерно наличие гипохромной микро-цитарной анемии, отмечается анизоцитоз эритроцитов. При оценке мазка крови обращает на себя внимание бледность эритроцитов, встречаются эритроциты в виде колец с широким просветлением в центре (анулоциты). При глубокой анемии отмечается выраженный анизоцитоз и пойкилоцитоз эритроцитов, могут появляться единичные мишеневидные клетки. Количество ретикулоцитов обычно в норме, т.к. регенераторная способность эритроидного ростка костного мозга сохранена. Транзиторный ретикулоцитоз может наблюдаться при выраженной кровопотере или при приеме препаратов железа незадолго до проведения анализов. У отдельных больных возможна умеренная лейкопения и может отмечаться тромбоцитопения (чаще у детей) или тромбоцитоз.
ЖДА диагностируется при сниженном уровне сывороточного железа ( 69 мкмоль/л (ОЖСС). Процент насыщения трансферрина железом (Кнас) 100 фл).
Заболевание обусловлено малъабсорбцией В/2 в результате атрофического гастрита и отсутствия секреции внутреннего желудочного фактора (перни-циозная анемия, болезнь Аддисона-Бирмера), гастрэктомия; алиментарный дефицит (в частности, у вегетарианцев); иногда болезни терминального отдела подвздошной кишки (болезнь Крона) или ее резекция; слепая петля; дивертикул; глистные инвазии (ВурМНоЪо1гшгп).
В12 содержится в печени и всех животных продуктах. Имеются запасы витамина в организме.
Нередко В12-ДА ассоциирована с заболеваниями щитовидной железы (до 25%), витилиго, болезнью Аддисона, карциномой желудка.
В клинической картине заболевания наряду с общеанемическими симптомами могут присутствовать признаки поражения центральной и периферической нервной системы, ЖКТ, что проявляется такими нарушениями, как: парестезии; периферическая нейропатия, нарушение позиционной и вибрационной чувствительности; нейропсихиатрические отклонения; глоссит — болезненный красный язык; диаррея.
Важно отметить, что неврологическая симптоматика (симптоматика так называемого «фуникулярного миелоза») может опережать развитие анемии.
Возможна умеренная желтушиость (лимонный оттенок кожи), умеренная спленомегалия и билирубинемия за счет непрямой фракции в связи с гемолизом (преимущественно внутрикостномозговым), нередко сопровождающим В12-ДА. Диагностика. Основное значение в диагностике ВП-ДА принадлежит морфологическим исследованиям крови и костного мозга. Анемия носит характер макроцитарной нормо- или гиперхромной, гипорегенераторной анемии. Отмечается анизо-, пойкилоцитоз, базофильная зернистость эритроцитов за счет наличия элементов РНК. В эритроцитах обнаруживаются остатки ядра в виде телец Жолли, колец Кебота. В клиническом анализе крови могут быть лейко-и тромбоцитопения, обычно умеренные, а также морфологические изменения гранулоцитов и тромбоцитов (формы больших размеров, гиперсегментация ядер нейтрофилов). Для уточнения диагноза показаны дополнительные исследования, включая исследование костного мозга для подтверждения мегалобластоид-ного типа кроветворения.
Имеются методы определения концентрации В]2 в сыворотке крови, что служит отражением запасов кобаламина в организме. Указанием на клинически значимый дефицит витамина Вр является его существенно сниженный сывороточный уровень.
У части больных могут обнаруживаться антитела к париетальным клеткам желудка или антитела к внутреннему фактору (специфические для пернициоз-ной анемии). В таких случаях иногда информативен тест Шиллинга, который назначается для определения, является ли дефицит В12 следствием мальабсорбции или отсутствия внутреннего фактора путем сравнения пропорции содержания в оральной дозе (1 мкг) радиоактивного В|2 с экскретируемым с мочой — при и без дополнительного назначения внутреннего фактора. Концентрация гомоци-стеина у больных с дефицитом В12 и дефицитом фолатов повышена.
Дифференциальная диагностика проводится с другими видами анемий, в первую очередь — макроцитарных, а также с фолиеводефицитной анемией. Понятие макроцитарной анемии отражает увеличенный размер эритроцитов, причиной которого могут быть нарушения, не связанные с синтезом ДНК. Дефицит витамина В12 следует отличать от таких заболеваний, как апластическая анемия, рефрактерная анемия или миелодиспластический синдром (МДС). Макроцитар-ную анемию с панцитопенией могут вызывать как гипо- так и гипертиреоидизм, а также алкоголизм, хронические заболевания печени. Причиной макроцитоза могут стать хронические заболевания почек и курение. Большое число ретику-лоцитов может повышать показатель МСУ, поскольку ретикулоциты являются крупными клетками. Вследствие этого гемолитическая анемия иногда ошибочно принимается за мегалобластную. В сложных случаях основным методом исследования является исследование костного мозга.
В лечении Вр-ДА важным моментом является устранение причины дефицита. Проводится заместительная терапия цианкобаламином до нормализации гематологических показателей или при ЦНС симптомах — пока не завершится восстановление. Большинству больных требуется поддерживающая терапия умеренными дозами В[2 на протяжении длительного времени (до полугода), а при неустранимости причины дефицита — в течение жизни. Предвестником начала улучшения является значительный ретикулоцитоз (как правило, после 4-5 дней) — ретикулоцитарный криз. Поскольку гемопоэз активизируется в ходе лечения, может быть необходимо дополнительное назначение препаратов железа. В трансфузиях эритроцитарной массы обычно необходимости нет.
Возможно полное гематологическое и неврологическое восстановление.
Причины низких фолатов — бедная диета (например, у алкоголиков), повышение потребности в фолатах (беременность, гемолиз, дизеритропоэз, опухоли, длительный гемодиализ), мальабсорбция, особенно при заболеваниях кишечника, тропическая спру, медикаменты (ряд противосудорожных препаратов, антагонист фолатов — метатрексат и др).
Фолаты содержатся в зеленых овощах, фруктах, печени и синтезируется кишечными бактериями. Запасы в организме истощаются относительно быстро при недостаточном поступлении.
Клиническая картина аналогична В]2-ДА, за исключением неврологических нарушений. Дефицит фолатов у матери также связан с дефектами нервной трубки у плода.
Диагностика. Картина крови и костного мозга не отличается от таковой при В12-ДА.
Для диагностики и дифференциальной диагностики используется определение уровня фолатов и В12 в сыворотке, а также фолатов эритроцитов.
При смешанных В]2-фолиеводефицитных формах анемии или неверной диагностике ФДА назначение одних фолатов может способствовать проявлению или ухудшению течения подострой комбинированной дегенерации спинного мозга.
Лечение. При ФДА проводится заместительная терапия фолиевой кислотой в виде перорального препарата. Терапия проводится под контролем показателей гемограммы (уровень гемоглобина и эритроцитов, эритроцитарные параметры, появление ретикулоцитарного криза) до нормализации показателей красной крови. При невозможности полного устранения факторов, способствующих развитию дефицита фолатов, в дальнейшем проводятся профилактические курсы терапии.
Прогноз благоприятный при адекватном лечении анемии и устранении причины заболевания.
Профилактика фолиеводефицитной анемии
Первичные профилактические мероприятия включают в себя наблюдение за лицами из групп риска, коррекцию диеты и назначение профилактических доз фолиевой кислоты при заболеваниях и состояниях, способствующих развитию ФДА. В частности, группу риска составляют больные эпилепсией, поскольку противосудорожные препараты являются потенциальными индукторами печеночных ферментов, а увеличение их активности приводит к ускоренному распаду фолатов и возникновению фолиеводефицитной мегалобластной анемии. Поэтому больных эпилепсией и пациентов, принимающих препараты из группы антиметаболитов, таких как метатрексат, необходимо регулярно обследовать для своевременного обнаружения анемии и проведения соответствующих мероприятий.
Гемолизом называют преждевременное разрушение эритроцитов. Он может происходить непосредственно в циркуляции (внутрисосудистый гемолиз) или в ретикулоэндотелиалъной системе (внесосудистый).
Причины гемолиза могут быть как генетически обусловленными, так и приобретенными. Генетические:
- Патология мембраны: врожденный сфероцитоз, эллиптоцитоз.
- Патология гемоглобина: серповидноклеточная болезнь — серповиднокле-точная анемия (СКВ = СКА), талассемия.
- Энзимные дефекты: дефицит глюкозо-фосфат дегидрогеназы (Г6 ФД), дефицит пируваткиназы и др.
- Иммунные: либо изоиммунные (гемолитическая болезнь новорожденных, посттрансфузионные реакции реакции гемолитического типа), аутоиммунные (обусловленные тепловыми или Холодовыми антителами), лекарственно-индуцированные.
- Неиммунные: травматические (кардиальный гемолиз, микроангиопатиче-ская анемия), инфекционные (малярия, септицемия), патология мембраны (па-роксизмальная ночная гемоглобинурия), заболевания печени.
Признаки гемолиза:
- Клинические: желтушность кожи, потемнение мочи, гепатоспленомегалия и др.
- Лабораторные:
— Связанные с повышенным разрушением эритроцитов:
— Повышение уровня билирубина (неконъюгированного);
— Увеличение содержания уробилина в моче;
— Снижение уровня гаптоглобина в сыворотке (связывает свободный гемоглобин).
— Связанные с повышенной продукцией эритроцитов:
— Гиперплазия костного мозга с расширением эритроидного ростка.
При установлении диагноза и проведении дифференциальной диагностики у больных гемолитическими анемиями необходимо обратить внимание на данные анамнеза (семейная история, национальная принадлежность, желтухи, гематурия, прием препаратов, ранее выявлявшейся анемии), на желтушность, гепатоспленомегалию, костные деформации (стигмы при наследственной патологии, особенности черепа при талассемии и др.), язвы на ногах (наблюдаются при СКВ, иногда при сфероцитозе).
Из лабораторных исследований показательными являются общий анализ крови с ретикулоцитами, уровень билирубина и фракционный его состав, ЛДГ, гаптоглобин (снижение уровня — показатель внутрисосудистого гемолиза), уро-билиноген мочи. Мазки крови могут показать полихромазию, макроцитоз, сфероцитоз, эллиптоцитоз, фрагментированные или серповидные клетки, мишене-видные клетки (характерны для талассемии). На следующем этапе проводятся специальные исследования, такие как тест Кумбса, определение гемосидерина мочи (индикатор хронического внутрисосудистого гемолиза). Аномалии мембраны могут быть подтверждены тестами на осмотическую стойкость. Электрофорез гемоглобина определяет варианты гемоглобина. Когда другие причины исключены, проводятся исследования ферментов.
Аутоиммунная гемолитическая анемия (АИГА) — анемия, при которой укорочение длительности жизни эритроцитов является результатом воздействия аутоантител против антигенов (мембранных белков) эритроцитов.
Частота встречаемости — около 1:100 000 населения.
Гемолиз может быть обусловлен тепловыми или Холодовыми антителами.
АИГА может быть самостоятельным заболеванием или выявляться при системных заболеваниях соединительной ткани, патологии щитовидной железы, синдроме Фишера—Эванса (нарушение иммунной регуляции с иммунной тромбоцито-лейкопенией, анемией в сочетании с рядом других нарушений). Известна ВИЧ-ассоциированная АИГА, вторичные АИГА вследствие микоплазменных, пневмококковых инфекций. Возможно появление аутоантител в результате повторных гемотрансфузий, беременностей. Холодовые агглютинины могут продуцироваться микоплазмой и ЭБВ.
Выделяют острую и хроническую формы. Для большинства случаев характерно острое начало с возможным переходом в хроническую форму. В зависимости от серологического варианта различают АИГА с полными и неполными антителами, с тепловыми и Холодовыми антителами, гемолизиновые формы.
Пароксизмальная холодовая гемоглобинурия (синдром Доната-Ландштей-нера), как правило, наблюдается после перенесенных вирусных инфекций и в поздних стадиях сифилиса.
В клинической картине
сочетаются симптомы анемии и гемолиза: потемнение мочи, иктеричность кожи и склер, лихорадка, боли в животе, умеренная гаштоспленомегалия. Особенностью холодовой АИГА является обострение хронической анемии на холоде, частое сочетание с синдромом Рейно или акро-цианозом. Гемолизиновые формы нередко сопровождаются гемоглобинурией и другими признаками острого внутрисосудистого гемолиза.
Диагностика.
Анемия, как правило, нормохромная нормоцитарная, характерен ретикулоцитоз, часто выраженный. Могут встречаться сфероциты в небольшом количестве. Возможен лейкоцитоз со сдвигом лейкоцитарной формулы плево, умеренный тромбоцитоз. Характерно повышение непрямого билирубина, эритроидная гиперплазия костного мозга. Увеличивается уровень лактатдеги-дрогеначы (ЛДГ) сыворотки. Уровень сывороточного железа в норме или повышен, гаптоглобина — в норме или снижен.
Дифференциальная диагностика
проводится с другими видами анемий, в первую очередь — гемолитическими, вторичными анемиями, болезнью Жиль-бера. Задача врачей общей практики — заподозрить данный вид анемии и провести первичную диагностику. Уточнение варианта и лечение обычно проводится в специализированных учреждениях.
Основным диагностическими тестом является положительный прямой анти-глобулиновый тест (проба Кумбса), определяющий антитела или комплемент на поверхности эритроцитов. Дополнительно проводится непрямой тест Кумбса, определяющий антитела в сыворотке.
В лечении аутоиммунных форм гемолитической анемии основное место принадлежит глюкокортикостероидам (ГК). У пациентов с острым гемолизом может использоваться внутривенный иммуноглобулин, обычно в сочетании с ГК.
При отсутствии эффекта от консервативной терапии возможно проведение спленэктомии, эффективность которой при данной патологии составляет около 70%. Из иммуносупрессивных препаратов при неэффективности обычной терапии в лечении АИГА используется азатиаприн, цитостатики (винкаалкалоиды, циклофосфамид), циклоспорин А.
Основой лечения симптоматических анемий является лечение базового заболевания.
Профилактика аутоиммунной гемолитической анемии
Первичные профилактические мероприятия заключаются в лечении основных заболеваний, при которых может возникнуть АИГА.
Вторичная прфилактика. Пациентам, страдающим АИГА для предотвращения усиления гемолиза и развития гемолитических кризов рекомендуется избегать провоцирующих факторов: переохлаждения при Холодовых формах, вирусных инфекций — при всех вариантах заболевания и др. Пациентам, которым произведена спленэктомия, учитывая развитие иммунодефицита, показано введение пневмококковой вакцины. Эта рекомендация в первую очередь относится к детям и лицам, имеющим дополнительные показания к вакцинации (по эпидемиологической обстановке и др.).
Наследственный сфероцитоз (НС) — цитоскелетная аномалия, обусловленная нарушением структуры спектрина. Результатом таких аномалий является потеря способности эритроцитов к деформации, нарушается работа Ш+/К+ — насоса мембраны, происходит преждевременная (не по мере старения) сферуляция эритроцитов, укорочение продолжительности жизни красных кровяных клеток и разрушение их клетками селезенки. Длительность жизни эритроцитов укорачивается до 12-14 дней.
Обусловлено мутациями в генах, кодирующих мембранные белки цитоске-лета эритроцитов. Наследование аутосомно-доминантное (проявляется анемией легкой и средней степени тяжести) или рецессивное (клинически проявляется в тяжелой форме).
Характеризуется гемолитической анемией, спленомегалией и наличием эритроцитов сферической формы в периферической крови. Болезнь может протекать скрыто.
НС основывается на наличии у пациента характерных морфологических изменений эритроцитов и признаков гемолиза. Показатели насыщения эритроцитов гемоглобином и уровень сывороточного железа обычно в норме, за исключением тех случаев, когда на фоне длительно существующего гемолиза в организме развивается железодефицитное состояние.
В костном мозге отмечается компенсаторное усиление эритропоэза.
Дифференциальный диагноз
проводится с желтухами другой этиологии (инфекционным гепатитом, обструктивной желтухой, синдромом Жильбера и др.), иммунной гемолитической анемией, микроангиопатической гемолитической анемией, спленомегалиями другой этиологии. При дифференциальной диагностике наряду с выявлением морфологически измененных эритроцитов, отрицательной пробой Кумбса и другими лабораторными данными, немаловажное значение может иметь тщательно собранный семейный анамнез и обследование родственников больного для выявления у них признаков НС.
При клинически компенсированном состоянии больного, отсутствии значимого гемолиза и анемии терапия обычно ограничивается симптоматическими средствами, в том числе направленными на профилактику развития желчекаменной болезни (желчегонные, фитотерапия, рациональная диета). При тяжелом гемолизе с выраженной анемией и при апластических кризах с низким уровнем гемоглобина производятся трансфузии эритроцитарной массы.
Одним методом терапии у больных сфероцитарной анемией является спле-нэктомия. Оперативное лечение показано больным с наличием гемолитической анемии средней и тяжелой степени или ее осложнений, в том числе при наличии желчнокаменной болезни, особенно у лиц молодого возраста. В результате удаления селезенки прекращается или значительно уменьшается гемолиз эритроцитов, увеличивается продолжительность их жизни.
Первичная профилактика
при НС, как и при других наследственных заболеваниях, заключается в генетическом консультировании и планировании семьи.
Вторичная профилактика.
Поскольку у значительной части пациентов заболевание протекает в скрытой или клинически компенсированной форме, то основные мероприятия по вторичной профилактике направлены на устранение проявлений хронической интоксикации, компенсацию повышенного расхода необходимых для кроветворения веществ и предупреждение таких осложнений, как раннее развитие желчнокаменной болезни. В связи с этим показано полноценное питание, прием поливитаминов с микроэлементами, желчегонных средств, ежегодный УЗИ-контроль состояния желчевыводящих путей.
Как и при других формах хронических гемолитических анемий у больных НС нередко развивается дефицит фолатов, в связи с чем этой категории больных профилактически назначается фолиевая кислота.
Анемии могут быть обусловлены подавленным (гипотастическим) состоянием кроветворения за счет токсических и радиационных воздействий, развития реактивного фиброза в костном мозге при ряде заболеваний или в результате самостоятельных заболеваний — гипопластической (апластической) анемии, парциальной красноклеточной аплазии.
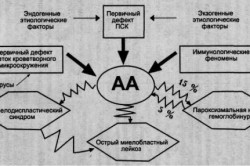
Апластическая анемия — тяжелое заболевание кроветворной системы, которое характеризуется панцитопенией в периферической крови и гипоклеточ-ным костным мозгом.
Заболевание редкое: от 2-3 до 10-20 случаев на млн населения в год. Наблюдается во всех возрастных группах. Большая частота встречаемости заболевания отмечается на Дальнем Востоке, в Японии, Таиланде.
Причинами развития заболевания могут быть цитотоксические препараты, радиация, лекарственные препараты (золото, хлорамфеникол), промышленные токсины, вирусы (гепатит). Этиологический фактор в половине случаев не выявляется — идиопатические формы. Выделяют врожденную форму — анемию Фанкони — генетически обусловленное заболевание с гиперчувствительностью к ДНК-повреждающим воздействиям и повышенной склонностью к развитию опухолевых заболеваний.
Современная концепция патогенеза АА предполагает связь между развитием аплазии кроветворения и дефектом стволовых клеток с нарушением их проли-феративной активности с участием иммуноопосредованных механизмов, нарушением регуляции гемопоэза иммунокомпетентными лимфоидными клетками.
Выделяют острую и хроническую формы заболевания, а также тяжелую апластическую анемию (тАА) и АА умеренной степени тяжести (нетяжелая апластическая анемия — нАА). ТАА определяется при наличии 2 любых из перечисленных критериев по данным периферической крови:
- Гранулоцитов менее 0,5 х 109/л
- Тромбоцитов менее 20 х 109/л
- Ретикулоцитов менее 1% (с коррекцией по гематокриту) в сочетании с аплазией костного мозга по данным трепанобиоптатов (клеточность костного мозга не более 30% от нормы).
Клинические проявления
заболевания обусловлены анемическим и геморрагическим синдромом.
Диагноз ставится на основании выявления характерных изменений в анализах крови и костном мозге с отсутствием признаков клонального гемопоэза. Осноиой диагностики является гистологическое исследование костного мозга.
Анемия нормохромного характера, количество ретикулоцитов снижено, как проянлспие I шюрегенераторного характера анемии.
И мпелограмме снижено количество ядросодержащих элементов, снижено суммарное процентное содержание клеточных элементов гранулопоэза и эритропоэза, нередко отмечается высокое относительное число лимфоцитов, значительно уменьшено содержание мегакариоцитов. В гистологических препаратах трепанопрепаратов подвздошной кости выявляется аплазия костного мозга с замещением кроветворной ткани жировой.
Содержание железа в сыворотке крови нормально или повышено.
Дифференциальный диагноз
проводится с гипопластическими вариантами гемобластозов (миелодиспластический синдром — МДС, острый лейкоз, су-блейкемический миелоз), вторичными — симптоматическими аплазиями, наблюдающимися при заболеваниях печени, ряде опухолевых заболеваний.
Пациентам с АА проводится иммуносупрессивная терапия, включающая глюкокортикоидные гормоны (ГК), антилимфоцитарный (АЛГ) или антитимоцитарный (АТГ) иммуноглобулин, циклоспорин-А (цА). Методом выбора в терапии тАА у пациентов до 40 лет является трансплантация костного мозга (ТКМ). Такая терапия позволяет получить ремиссии у 70 -80%. Проводится также симптоматическая терапия, направленная на коррекцию анемического и геморрагического синдромов, профилактику и лечение возможных инфекционных и иных осложнений.
Прогноз заболевания в первую очередь зависит от глубины аплазии и тяжести заболевания, а также своевременности и активности проводимой терапии.
Основные причины смерти больных — геморрагические и инфекционные осложнения, прогрессирование аплазии при безуспешной терапии.
Первичные профилактические мероприятия предусматривают прекращение контакта с факторами, обладающими гемодепрессивными свойствами, ограничение использования лекарственных препаратов с миелосупрессивными свойствами. Так, в ряде стран прекращено применение препарата левомецитин (хлорамфеникол), поскольку была показана связь приема данного лекарственного средства с повышением частоты развития аплазии кроветворения. При развитии АА на фоне беременности целесообразно ее прерывание.
Вторичная профилактика.
Пациенты с ремиссией заболевания должны оставаться под наблюдением с регулярным контролем показателей гемограммы, поскольку возможны рецидивы заболевания, как под воздействием неблагоприятных факторов, так и спонтанные.
источник
Отмечается эритропения, снижение содержания гемоглобина до 30-40 г/л, анизоцитоз, пойкилоцитоз, гипохромия, снижение ретикулоцитарного индекса, лейкопения за счет снижения количества нейтрофилов.
Патогномоничный признак железодефицита в костном мозге – резкое снижение местных запасов железа (снижается количество сидеробластов – клеток, содержащих гранулы железа). Отмечается умеренная гиперплазия эритроидного ростка. Гемоглобинизация клеток эритрона нарушена: увеличено количество базофильных и полихроматофильных форм, снижено количество оксифильных. По мере истощения запасов железа ферритин и гемосидерин исчезают и из костного мозга, и из других мест хранения.
Особую роль в диагностике играют биохимические показатели:
Уровень ферритина сыворотки снижен – менее 12 нг/мл.
Увеличена общая железосвязывающая способность.
Увеличена ненасыщенная, латентная железосвязывающая способность.
Резко снижено насыщение трансферрина.
Витамин В12 содержится в мясе, яйцах, сыре, молоке, почках, печени. Витамин В12 в продуктах связан с белком, от которого он освобождается в процессе кулинарной обработки и под воздействием протеолитических ферментов ЖКТ, после чего он связывается в желудке с внутренним фактором Кастла. Внутренний фактор – это гликопротеид, который образуется в фундальной части и в области тела желудка париетальными клетками. Комплекс «витамин В12 – фактор Кастла» связывается со специфическими рецепторами клеток подвздошной кишки. Витамин В12 всасывается медленно, за сутки может всосаться не более 6-9 мкг. Незначительная часть витамина В12 может всосаться без фактора Кастла.
Витамин В12 в плазме связывается с транскобаламинами. Известно три транскобаламина – I, II, III. Основное количество витамина переносится транскобаламином II. Этот белок синтезируется в печени.
Содержание витамина В12 в организме здорового человека составляет 2-5 мг, депо находится в печени. Запасы витамина В12 настолько велики, что их хватает на 3-6 лет.
Все клетки организма имеют рецепторы для комплекса «транскобаламин II – витамин В12».
У человека выявлены две метаболически активные формы витамина В12:
1. Метилкобаламин. Эта активная форма обеспечивает нормальное эритробластическое кроветворение и нормально протекающие метотические процессы в других клетках организма. В ходе реакции с метилкобаламином из уридинмонофосфата образуется тимидинмонофосфат, который включается в ДНК. Необходимым условием для течения этой реакции является участие активной коферментной формы фолиевой кислоты – 5, 10 – метилен-тетрагидрофолиевой кислоты.
Метилкобаламин участвует в метилировании гомоцистеина в метионин.
2. 5-дезоксиаденозилкобаламин. Этот метаболит необходим для нормального обмена жирных кислот. Так при распаде некоторых жирных кислот образуется пропионовая кислота. Распад пропионовой кислоты обеспечивается рядом ферментативных реакций, в ходе которых синтезируются производные янтарной кислоты, входящие в цикл Кребса. Одним из промежуточных продуктов является метилмалоновая кислота. Метилмалоновая кислота образуется также при распаде валина и метионина. 5-дезоксиаденозилкобаламин участвует в образовании янтарной кислоты из метилмалоновой. Предполагается участие аденозилкобаламина в биосинтезе миелина. При дефиците витамина В12 накапливается избыток пропионовой и метилмалоновой кислот. Они переходят в жирные кислоты с физиологическим нечетным числом атомов углерода в молекуле. Включаясь в липиды нейронов, они нарушают процессы миелинизации и вызывают жировую дистрофию клеток.
источник

Ретроспективными исследованиями установлено, что средний интервал от воздействия этиологического агента до возникновения панцитопении составляет 6-8 нед.
Симптомы апластических анемий напрямую связаны со степенью снижения 3 важнейших показателей периферической крови — гемоглобина, тромбоцитов и нейтрофилов. Подавляющее большинство больных апластическими анемиями обращаются к врачу по поводу кровоточивости, причём угрожающие жизни кровотечения в качестве первого клинического проявления болезни встречаются очень редко. В типичных случаях речь идёт о петехиальной сыпи, кровоточивости дёсен и легко возникающих экхимозах. Серьёзные висцеральные кровотечения — желудочно-кишечные, почечные и внутричерепные — возникают позже. Анемический синдром проявляется лёгкой утомляемостью, шумом в ушах, ощущением пульсации в голове, усталостью и другими классическими симптомами анемии. Как правило, дети хорошо переносят даже очень тяжёлую анемию. По данным литературы, тяжёлые инфекции редко выступают первыми симптомами болезни, однако, по нашим данным, это не совсем так. Нехарактерны для апластических анемий снижение массы, спленомегалия, лимфаденопатия и боли. Появление этих симптомов заставляет искать другую причину панцитопении.
Помимо внимательного клинического осмотра минимальный спектр необходимых диагностических исследований при подозрении на апластическую анемию включает:
- гемограмму с определением ретикулоцитов и ручным подсчётом лейкоцитарной формулы;
- миелограмму из 2-3 анатомически различных точек;
- трепанобиопсию костного мозга;
- пробу на ломкость хромосом с диэпоксибутаном или митомицином (митомицин С);
- биохимический анализ крови.
Для апластических анемий типично конкордатное снижение показателей производных всех 3 главных ростков костномозгового кроветворения (эритроцитов, гранулоцитов и тромбоцитов), несмотря на различную кинетику зрелых элементов крови. У большинства пациентов число лимфоцитов и моноцитов также снижено. Абсолютное число ретикулоцитов неадекватно тяжести анемии. Повышение фетального гемоглобина наряду с макроцитозом типично для апластических анемий. Повышение активности сывороточных трансаминаз, за исключением случаев гепатит-ассоциированных апластических анемий нехарактерно. Относительно высокий ретикулоцитоз, повышение билирубина и лактатдегидрогеназы говорит о сопутствующем синдроме — пароксизмальной ночной гемоглобинурии.
Состояние костного мозга при апластических анемиях должно быть оценено как по данным аспирата из нескольких точек, так и по данным трепанобиопсии. По данным исследования пунктата оценивают морфологию резидентных элементов эритро-, грануло- и мегакариоцитопоэза. Дизэритропоэз — весьма частая характеристика при апластических анемиях, типично также выявление «мегалобластоидности» эритроидных элементов, асинхронии созревания ядра и цитоплазмы эритробластов — эти признаки очень трудно дифференцировать от дисплазии эритроидного ряда, выявляемой при миелодиспластических синдромах. Зачастую в пунктате выявляется повышение количества плазматических клеток и макрофагов с явлениями фагоцитоза эритроцитов. Выявление лейкемических бластов в аспирате заставляет пересмотреть диагноз.
В 1976 и 1979 гг. Брюсом Камиттой и соавт. была выделена группа простейших показателей периферической крови и костного мозга, определяющих тяжесть течения заболевания и прогноз больных апластической анемией.
Критерии тяжёлой формы апластической анемии
Клеточность костного мозга, по данным трепанобиопсии, менее 25% (или 9 /л). Тромбоцитопения по мере прогрессирования заболевания достигает значительной степени (до единичных тромбоцитов в мазке). СОЭ, как правило, увеличена.
При анемии Фанкони имеется стресс-эритропоэз, который характеризуется макроцитозом, высоким уровнем Hb F, высоким уровнем эритропоэтина в сыворотке и наличием i-антигена.
Стернальный пунктат на ранних стадиях заболевания нормо- или гипоклеточный. Количество бластов в пределах нормы. Содержание клеток эритроидного ростка увеличено с задержкой их созревания и морфологическими нарушениями в виде анизоцитоза, базофильной пунктации в нормобластах, иногда отмечается появление мегалобластов. Гранулоцитарный росток «сужен», возможна задержка созревания на стадии неитрофильных миелоцитов и метамиелоцитов. Мегакариоцитарный росток значительно «сужен» уже на ранних стадиях заболевания. По мере прогрессирования заболевания отмечается выраженная гипоклеточность костного мозга с угнетением всех ростков и разрастанием жировой ткани. В костном мозге увеличено число ретикулярных, плазматических и тучных клеток. Гипоплазия костного мозга подтверждается результатами трепанобиопсии.
Из биохимических показателей для апластической анемии характерно повышение уровня фетального гемоглобина до 15 % (при норме 2 %) еще до развития цитопении, по мере прогрессирования аплазии фетальный гемоглобин достигает 45 %.
Установлено, что клетки больных с анемией Фанкони не способны репарировать поперечные сшивки ДНК, вызываемые так называемыми кластогенами — диэпоксибутаном, митомицином С и др. На этом феномене основана современная диагностика анемии Фанкони и у всех больных с подозрением на анемию Фанкони должен проводиться тест с диэпоксибутаном.
Течение анемии Фанкони характеризуется наличием периодов обострения и ремиссии. Без лечения через 2 года после диагностики панцитопении умирают 80 % больных, а через 4 года — около 100 %. Причиной смерти, наряду с тяжелой анемией, являются наиболее серьезные проявления геморрагического синдрома — желудочно-кишечные кровотечения, внутричерепные кровоизлияния и присоединение различных инфекций.
У больных анемией Фанкони имеется высокий риск трансформации в миелодиспластический синдром, острый лейкоз (особенно миелобластный или монобластный), злокачественные опухоли ЖКТ.
Наследственная апластическая анемия с общим поражением гемопоэза без врожденных аномалий развития (анемия Эстрена-Дамешека)
Является тотальной формой наследственной апластической анемии, наследуется аутосомно-рецессивно, протекает с панцитопенией, не сопровождается врожденными пороками развития. Заболевание встречается крайне редко, гематологические нарушения отмечаются в раннем детском возрасте. Прогноз неблагоприятный.
Врожденный дискератоз (синдром Цинссера-Коула-Энгмана)
Синдром характеризуется признаками эктодермальной дисплазии (патологическое ороговение отдельные клеток шиповатого слоя эпидермиса кожи и слизистых оболочек) в сочетании с гематологическими изменениями (примерно у 50 % больных развивается апластическая анемия). В 75 % случаев синдром наследуется рецессивно сцепленно с Х-хромосомой и, соответственно, встречается у мальчиков; у 25 % детей больных наследуется по аутосомно-доминантному типу (описано примерно одинаковое количество больных). Поражаются кожа и ее дериваты, слизистые оболочки. Наблюдаются множественный рассеянный гиперкератоз с преимущественной локализацией на лице, шее, спине, груди; атрофия кожи ладоней и ступней, ладонно-подошвенный гипергидроз; нарушение роста и дистрофия ногтей; гипотрихоз ресниц; закупорка слезных канальцев и слезотечение; лейкоплакия слизистых полости рта, в основном языка и десен; поражение эндокринных желез (нанизм, недоразвитие вторичных половых признаков). Гематологические изменения разнообразны: панцитопения, изолированная анемия, тромбоцитопения, нейтропения. Возраст дебюта апластической анемии при данном синдроме может быть весьма вариабелен, средний возраст дебюта АА составляет 15 лет.
В отличие от больных с анемией Фанкони клетки больных с врожденным дискератозом не обладают повышенной чувствительностью к антигенам, вызывающим поперечные сшивки, поэтому эти иногда фенотипически похожие синдромы могут быть дифференцированы на основании теста с диэпоксибутаном.
Характеризуется экзокринной недостаточностью поджелудочной железы, карликовостью, метафизарной хондродис-плазией, нейтропенией, иногда анемией, тромбоцитопенией. Наследуется аутосомно-доминантно.
Заболевание клинически проявляется в раннем возрасте и характеризуется признаками поражения ЖКТ и гематологическими изменениями. Отмечаются диарея, стеаторея, замедление весовых прибавок, гипотрофия. Характерны изменения костной системы в виде хондроднсплазии метафиза и формирование ортопедической патологии, задержка роста. У части больных возможна галактоземия, что приводит к гепатоспленомегалии, задержке психомоторного развития. Характерны рецидивирующие респираторные заболевания, отиты, абсцессы, остеомиелит. У некоторых детей наблюдается задержка наступления пубертатного периода.
В анализах крови с раннего возраста отмечается абсолютная нейтропения, число нейтрофилов менее 1 х 10 9 /л. Для зрелых нейтрофилов характерна гипосегментация ядер, отмечается снижение хемотаксиса нейтрофилов. Наряду с нейтропенией приблизительно у 50 % больных отмечается анемия с ретикулоцитопенией, у 60-70 % детей — тромбоцитопения, примерно у 25% больных развивается апластическая анемия. В стернальном пунктате число миелокариоцитов может быть нормальным, сниженным или повышенным; отмечается задержка созревания нейтрофилов на стадии метамиелоцита. Прогноз наиболее неблагоприятный в раннем детском возрасте, когда около 25% детей погибают от инфекционных осложнений; летальный исход возможен и от кровоизлияний в жизненно важные органы.
Наследственная апластическая анемия с избирательным поражением эритропоэза (анемия Блекфена-Даймонда)
Частота данного заболевания составляет 1 : 1 000 000 живых новорожденных; 5 — 7 : 1 000 000 во Франции, 10:1 000 000 в Скандинавии, встречается во всех этнических группах, мальчики и девочки заболевают одинаково часто. Подавляющее большинство (75 %) составляют спорадические случаи заболевания; в некоторых случаях возможно аутосомно-доминантное наследование, аутосомно-рецессивное или сцепленное с Х-хромосомой.
Первые признаки болезни выявляются в первые месяцы или на протяжении первого года жизни — у 35 % больных анемией при рождении, у 65 % в первые 6 месяцев жизни и в 90 % случаев заболевание диагностируется до года. Диагноз анемии Блекфена — Даймонда у детей старше 2 лет маловероятен. Дети, как правило, рождаются доношенными с нормальной массой тела и ростом, психомоторное развитие нормальное. Бледность кожи и слизистых отмечается с первых дней жизни, но явные клинические признаки гипоксии: вялость или возбуждение, беспокойство, сонливость, отказ от еды, диспепсические явления — появляются при снижении гемоглобина до 60-30 г/л. Врожденные пороки развития встречаются реже (в 25 % случаев), чем при анемии Фанкони. Некоторые больные имеют характерные фенотипические особенности: волосы цвета пакли, курносый нос, большая верхняя губа, гипертелоризм. По мере прогрессирования заболевания кожа приобретает восковидный, а к 5-6 годам, в связи с развитием гемосидероза, — сероватый оттенок, особенно в области шейных, подмышечных, паховых складок, половых органов. Геморрагический синдром отсутствует. Наблюдаются гепатомегалия, спленомегалия, в динамике заболевания селезенка сокращается, а печень прогрессивно увеличивается. Костный возраст отстает от паспортного на 4-5 лет, темпы окостенения изменены. Смена молочных зубов запаздывает, часто выявляется кариес.
В периферической крови нормохромная макроцитарная гипо- или арегенераторная анемия (ретикулоциты 0-0,1 %), как правило, тяжелой степени. Число лейкоцитов и тромбоцитов остается на нормальном уровне в течение первых лет жизни; иногда отмечается тенденция к тромбоцитозу. При длительном течении заболевания может развиваться умеренная тромбоцитопения. После первого десятилетия жизни может также появиться умеренная нейтропения, вероятно, из-за снижения клональной эффективности предшественников гранулоцитов.
Биохимически отмечается высокий уровень активности эритроцитарной аденозиндезаминазы; уровень фетального гемоглобина нормальный или умеренно повышен; повышено содержание i-антигена в эритроцитах; увеличено содержание эритропоэтина в сыворотке.
В стернальном пунктате костный мозг нормоклеточный, по мере прогрессирования заболевания отмечается гипоклеточность. Эритроидный росток резко сужен; диагностическим критерием является отсутствие или малое количество эритробластов (менее 5 % ядросодержащих клеток) в костном мозге. Миелоидный и мегакариоцитарный ростки не изменены. Число ретикулярных клеток и лимфоцитов увеличено, плазматических клеток не изменено.
Анемия Блекфена-Даймонда протекает хронически, у 80 % больных получают ремиссию при использовании кортикостероидов; у 20 % больных описана спонтанная ремиссия. «Постоянная гипоксия, нарушение утилизации железа, необходимость по жизненным показаниям трансфузии эритроцитарной массы неуклонно ведут к гемосидерозу, который в дальнейшем является «убийцей» больного ребенка». Возможна трансформация в миелодиспластический синдром, острый лейкоз (лимфобластный, миелобластный, промиелоцитарный, мегакариоцитарный), солидные опухоли (гепатобластому, рстеосаркому, злокачественную фиброзную гистиоцитому), лимфогранулематоз.
Дифференциальный диагноз при анемии Блекфена-Даймонда проводится с другими видами анемий, при которых в периферической крови уменьшается количество ретикулоцитов.
Анемия в период выздоровления после гемолитической болезни новорожденных.
Иногда может сочетаться со снижением интенсивности эритропоэза. Апластические кризы, характеризующиеся ретикулоцитопенией и уменьшением числа предшественников эритроцитов, могут осложнять разные типы гемолитической болезни. Подобные эпизоды являются транзиторными, кроме того, обычно выявляются признаки предшествующей гемолитической болезни. Развитие апластических кризов связывают с В19-парвовирусной инфекцией. Тактика ведения больных, как правило, выжидательная: при значительном снижении уровня гемоглобина проводятся гемотрансфузии.
Транзиторная эритробластопения детского возраста
Одна из наиболее частых форм аплазии эритроидного ростка. Этиология заболевания не известна. У здоровых ранее детей в возрасте 5 мес — 6 лет, чаще всего в возрасте 2 лет, медленно развивается тяжелая арегенераторная анемия, обусловленная резким снижением эритроцитов в костном мозге.
Развитию анемии за 1 — 2 мес может предшествовать вирусная инфекция, хотя связь заболевания с определенным возбудителем не доказана, нередко им является парвовирус В19. Анамнез и физикальное исследование неинформативны, обращает на себя внимание лишь выраженная бледность кожных покровов и слизистых. В периферической крови уровень Нb снижен до 30-80 г/л, ретикулоциты отсутствуют, количество лейкоцитов и тромбоцитов обычно нормальное, однако у 10 % больных имеется нейтропения ( 9 /л) и у 5 % — тромбоцитопения ( 9 /л). Лабораторно выявляют нормальные уровни активности эритроцитарнои аденозиндезаминазы и фетального гемоглобина; по ферментативным характеристикам эритроциты относят к стареющей популяции. Уровень железа в сыворотке повышен. В пользу транзиторной эритробластопении также свидетельствуют нормальные результаты клинического анализа крови до болезни. В стернальном пунктате отмечается резкое сужение эритроидного ростка, отсутствуют предшественники, за исключением нормоцитов, эритроцитов. Культуральными исследованиями костного мозга выявлено несколько патогенетических механизмов: присутствие в сыворотке ингибиторов стволовых клеток или аномалии последних, выражающиеся либо в их численности, либо в способности к реакции на эритропоэтин. Возможен аутоиммунный генез заболевания с поражением первичных эритроидных предшественников, а не зрелых эритроцитов. Через несколько месяцев после старта заболевания наступает спонтанная ремиссия. До наступления выздоровления могут потребоваться гемотрансфузии, кортикостероиды не используются.
Вторичные (приобретенные) аплазии эритроидного ростка
Также проявляются анемией, сопровождающейся ретикулоцитопенией и уменьшением количества предшественников эритроцитов в костном мозге. Вторичная аплазия эритроидного ростка может быть вызвана вирусными инфекциями (паротит, вирус Эпштейна-Барр, парвовирус В19), а т и — пичными пневмониями и бактериальным сепсисом; лекарственными средствами (хлорамфеникол, пенициллин, фенобарбитал, дифенилгидантоин); антиэритроцитарными антителами; иммунодефицитом; тимомой; злокачественными опухолями.
Эпизоды острой недостаточности эритропоэза могут сопровождать целый ряд вирусных инфекций. При этом значительно снижается количество циркулирующих ретикулоцитов (менее 0,1 %) и повышается уровень железа в сыворотке. В костном мозге уменьшено число предшественников эритроцитов. Эти эпизоды, как правило, купируются и не оставляют каких-либо последствий. Наиболее часто вторичная аплазия эритроидного ростка вызывается парвовирусом В19.
У всех больных грудного возраста при диагностике эритробластопении необходимы следующие исследования:
- Содержание сывороточных антител IgM и IgG (мать и ребенок).
- Вирусной ДНК в сыворотке крови.
- Вирусной ДНК в костном мозге.
Эти исследования могут помочь в дифференциации эритробластопении при инфицировании парвовирусом В19 и эритробластопении другого генеза.
В терапии вторичных эритробластопении важно устранение причины, вызвавшей заболевание — отмена препарата, лечение основного заболевания или тимэктомия. При обнаружении антиэритроидных антител показаны кортикостероиды, при их неэффективности — иммунодепрессанты (циклофосфан или азатиоприн). При иммунодефиците парвовирусная инфекция может быть хронической, тогда применяют иммуноглобулин внутривенно.
Приобретенные апластические анемии
Клиника приобретенных апластических анемий отличается в зависимости от тотального или избирательного поражения гемопоэза. У больных приобретенными апластическими анемиями в отличие от наследственных форм отсутствуют врожденные аномалии развития, физическое и психическое развитие детей не изменено, костный возраст соответствует паспортному.
Для тотальных форм апластических анемий характерно сочетание геморрагического, анемического и инфекционно-септического синдромов. Геморрагический синдром, обусловленный тромбоцитопенией, выражен резко: множественные экхимозы и петехии на коже и слизистых оболочках, конъюнктивах, рецидивирующие носовые, десневые, маточные, желудочно-кишечные и почечные кровотечения, кровоизлияния в местах инъекций. Непосредственной причиной смерти у таких больных являются чаще всего кровоизлияния в жизненно важные органы. Поражение эритроидного ростка приводит к развитию анемического синдрома, при котором у больного отмечается общая слабость, снижение аппетита, головокружение, повышенная утомляемость, бледность кожи и слизистых оболочек, ногтевых фаланг, изменения со стороны сердечно-сосудистой системы: расширение границ сердца, приглушенность тонов, тахикардия, систолический шум различной интенсивности, возможна экстрасистолия, одышка. Наличие лейкогранулоцитопении обусловливает возникновение инфекционно-септического синдрома: легкое присоединение инфекций любой локализации, язвенно-некротическое поражение кожи, слизистых оболочек. Характерно тяжелое течение инфекций, вызванных не только патогенной флорой, но и условно-патогенными и грибковыми возбудителями. Лимфатические узлы, печень, селезенка не увеличены. При избирательном поражении эритроидного ростка имеются проявления лишь анемического синдрома.
Все симптомы болезни могут проявляться и нарастать более или менее остро.
Гематологические изменения при апластической анемии складываются из нейтропении (абсолютное число нейтрофилов менее 1,5 х 10 9 /л), анемии (Нb 9 /л) и ретикулоцитопении, не соответствующей тяжести анемии. В миелограмме отмечается резкое снижение клеточности, редукция миелоидного и эритроидного ростков, вариабельный лимфоцитоз и отсутствие мегакариоцитов. У больных с медленным развитием аплазии длительно могут сохраняться участки активного кроветворения — «горячие карманы». В трепанобиоптате обнаруживается резкое снижение плацдарма кроветворения — доминирует жировой костный мозг, гемопоэтические элементы представлены резидуальными очагами эритро- и миелопоэза, мегакариоциты практически не обнаруживаются.
По тяжести приобретенные апластические анемии делятся в зависимости от глубины цитопении, ретикулоцитоза и остаточной клеточности костного мозга по данным трепанобиопсии. Используются критерии тяжести апластической анемии, разработанные международной группой изучения апластической анемии — «критерии Камитты»:
- число гранулоцитов менее 500 в 1 мкл;
- число тромбоцитов менее 20 000 в 1 мкл;
- число ретикулоцитов менее 40 000 в 1 мкл (или менее 1 % после коррекции на нормальный гематокрит).
Апластическая анемия считается тяжелой, если присутствуют любые два указанных выше показателя крови в сочетании со сниженной клеточностью. Если гематологический синдром соответствует критериям тяжелой апластической анемии, но число гранулоцитов менее 200 в 1 мкл — сверхтяжелая апластическая анемия. Все остальные случаи характеризуются как нетяжелая апластическая анемия.
Дифференциальный диагноз при приобретенных апластических анемий проводится преимущественно с острым лейкозом, мегалобластными анемиями, синдромом гиперспленизма, метастазами опухолей в костный мозг.
 [1], [2], [3], [4], [5], [6], [7], [8]
[1], [2], [3], [4], [5], [6], [7], [8]
источник