РЦРЗ (Республиканский центр развития здравоохранения МЗ РК)
Версия: Клинические протоколы МЗ РК — 2017
| МКБ-10 | |
| Код | Название |
| D50 | Железодефицитная анемия |
| D50.0 | Постгеморрагическая (хроническая) анемия |
| D50.8 | Другие железодефицитные анемии |
| D50.9 | Железодефицитная анемия неуточненная |
Дата разработки/пересмотра протокола: 2013 год (пересмотрен в 2017 г.)
Сокращения, используемые в протоколе:
(англ. Iron-refractory iron-deficiency anemia) железорефрактерная железодефицитная анемия
(англ. Mean corpuscular hemoglobin) – среднее содержание гемоглобина в эритроците в пг
(англ. Mean corpuscular volume) средний объем эритроцита в фл
сатурация трансферрина (англ. Transferrin Saturation) или коэффициент насыщение трансферрина железом
общая железосвязывающая способность сыворотки
растворимые рецепторы трансферрина
Пользователи протокола: врач общей практики, терапевт, гематолог, гинеколог, хирург, ревматолог, нефролог, пульмонолог, гастроэнтеролог, эндокринолог.
Категория пациентов: взрослые.
Шкала уровня доказательности:
| А | Высококачественный мета-анализ, систематический обзор РКИ или крупное РКИ с очень низкой вероятностью (++) систематической ошибки результаты, которых могут быть распространены на соответствующую популяцию. |
| В | Высококачественный (++) систематический обзор когортных или исследований случай-контроль или Высококачественное (++) когортное или исследований случай-контроль с очень низким риском систематической ошибки или РКИ с невысоким (+) риском систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
| С | Когортное или исследование случай-контроль или контролируемое исследование без рандомизации с невысоким риском систематической ошибки (+). Результаты, которых могут быть распространены на соответствующую популяцию или РКИ с очень низким или невысоким риском систематической ошибки (++ или +), результаты которых не могут быть непосредственно распространены на соответствующую популяцию. |
| D | Описание серии случаев или неконтролируемое исследование или мнение экспертов. |
| GPP | Наилучшая клиническая практика. |
Классификация[1,3]:
Общепризнанной классификации ЖДА не существует.
Латентный дефицит железа, функциональный дефицит железа, анемия хронических заболеваний являются самостоятельными нозологическими формами в классификацию ЖДА не включены.
Наиболее часто используется классификация по степени тяжести и этиологическая классификация [1].
Классификация ЖДА по степени тяжести [3]:
Этиологическая классификация ЖДА (по Camaschella C., 2015 в модификации)[1]:
| Причина | Примеры |
| Повышенное потребление железа | Быстрый рост в подростковом периоде, менструальные кровопотери, беременность во втором и третьем триместрах, донорство крови |
| Недостаточное алиментарное поступление железа | Недостаточное алиментарное поступление железа вследствие недоедания, обусловленного социальными причинами, вегетарианства и др. |
| Нарушение абсорбции железа | Гастроэктомия, дуоденальный шунт, бариатрическая хирургия, целиакия, воспалительные заболевания кишечника, атрофический гастрит, глистная инвазия |
| Хронические кровопотери | Из желудочно-кишечного тракта: эзофагит, гастрит, язва желудка, язва двенадцатиперстной кишки, дивертикулез, опухоли желудочно-кишечного тракта, воспалительные заболевания кишечника, ангиодисплазия, геморрой, паразитоз, оккультные кровотечения Из половых и мочевыводящих путей: обильные и/или продолжительные менструации, внутрисосудистый гемолиз (в т.ч. при пароксизмальной ночной гемоглобинурии, аутоиммунной гемолитической анемии с холодовыми антителами, маршевая гемоглобинурия, микроангиопатический гемолиз, повреждение эритроцитов протезом клапанов) Системные кровотечения, включая геморрагическую телеангиоэктазию, хронический шистосомоз, синдром Мюнхгаузена |
| Связанные с лекарственными препаратами | Глюкокортикостероиды, салицилаты, нестероидные противовоспалительные, ингибиторы протонной помпы [4, 5] |
| Наследственные | IRIDA (мутация в гене TMPRSS6) и более редкие причины |
| Эритропоэз, ограниченный железом | Лечение с использованием эритропоэтинов анемии хронических заболеваний, хронической болезни почек |
МЕТОДЫ, ПОДХОДЫ И ПРОЦЕДУРЫ ДИАГНОСТИКИ [1-3]
Диагностические критерии
Жалобы и физикальное обследование:
Общеанемический синдром: слабость, повышенная утомляемость, головокружение, головные боли (чаще в вечернее время), одышка при физической нагрузке, ощущение сердцебиения, синкопальные состояния, мелькание «мушек» перед глазами при невысоком уровне артериального давления, часто наблюдается умеренное повышение температуры, нередко сонливость днем и плохое засыпание ночью, раздражительность, нервность, конфликтность, плаксивость, снижение памяти и внимания, ухудшение аппетита. Выраженность жалоб зависит от адаптации к анемии. Лучшей адаптации способствует медленный темп анемизации.
Сидеропенический синдром:
· изменение кожи и ее придатков (сухость, шелушение, легкое образование трещин, бледность). Волосы тусклые, ломкие, «секутся», рано седеют, усиленно выпадают, изменения ногтей: истончение, ломкость, поперечная исчерченность, иногда ложкообразная вогнутость (койлонихии).
· изменения слизистых оболочек (глоссит с атрофией сосочков, трещины в углах рта, ангулярный стоматит).
· изменения со стороны желудочно-кишечного тракта (атрофический гастрит, атрофия слизистой пищевода, дисфагия). Затруднение глотания сухой и твердой пищи.
· мышечная система. Миастения (вследствие ослабления сфинктеров появляются императивные позывы на мочеиспускание, невозможность удерживать мочу при смехе, кашле, иногда ночное недержание мочи у девочек). Следствием миастении могут быть и невынашивание беременности, осложнения в процессе беременности и родов (снижение сократительной способности миометрия).
· пристрастие к необычным запахам.
· извращение вкуса. Выражается в стремлении есть что-либо малосъедобное.
· склонность к тахикардии, гипотонии.
Лабораторные исследования:
Лабораторные исследования при подозрении на ЖДА могут включать помимо ОАК с ретикулоцитами и показателей обмена железа также исследование уровня витамина В12, фолиевой кислоты, биохимические показатели (общий белок, креатинин, мочевина, глюкоза крови, общий билирубин, прямой билирубин, трансаминазы) и другие исследования в зависимости от особенностей клинической картины и широты дифференциально-диагностического поиска. План исследований также может расширяться для уточнения причин железодефицита и исключения скрытых кровопотерь и проведения онкопоиска.
| № п/п | Лабораторный показатель | Референсный интервал (может меняться в зависимости от лаборатории) | Изменения при ЖДА |
| 1 | Морфологические изменения эритроцитов | нормоциты – 68% микроциты – 15,2% макроциты – 16,8% | Микроцитоз сочетается с анизоцитозом, пойкилоцитозом, в наличии анулоциты, плантоциты |
| 2 | Цветовой показатель | 0,86 -1,05 | Гипохромия показатель менее 0,86 |
| 3 | Содержание гемоглобина | Женщины – не менее 120 г/л Мужчины – не менее 130 г/л | Уменьшено |
| 4 | МСН | 27-31 пг | Менее 27 пг |
| 5 | МСНС | 33-37% | Менее 33 % |
| 6 | МСV | 80-100 фл | Снижен |
| 9 | Количество ретикулоцитов | 2-10:1000 | Не изменено |
| 11 | Железо сыворотки | Женщины – 12-25 мкмль/л Мужчины –13-30 мкмоль/л | Снижено |
| 12 | Общая железосвязывающая способность сыворотки крови | 30-85 мкмоль/л | Повышена |
| 13 | Латентная железосвязывающая способность сыворотки | Менее 47 мкмоль/л | Выше 47 мкмоль/л |
| 14 | Коэффициент насыщения трансферрина железом (TSat) | ≥16% | Уменьшено |
| 15 | Уровень ферритина * | 15-150 мкг/л | Уменьшение |
* — показатель информативен только при отсутствии признаков системного воспалительного ответа (например — при нормальном уровне СРБ)
Инструментальные исследования:
С целью выявления источников кровопотери, патологии других органов и систем, в том числе солидных опухолей:
· фиброгастродуоденоскопия по показаниям;
· рентгенологическое исследование органов ЖКТ по показаниям;
· рентгенологическое исследование органов грудной клетки по показаниям;
· фиброколоноскопия по показаниям;
· ректороманоскопия по показаниям;
· УЗИ органов малого таза по показаниям;
· УЗИ органов брюшной полости по показаниям;
Показания для консультации специалистов:
· консультация хирурга – для исключения кровотечения из желудочно-кишечного тракта;
· консультация гастроэнтеролога – при подозрении на мальабсорбцию или хронические кровопотери при патологии органов желудочно-кишечного тракта;
· консультация стоматолога – стоматологические проблемы, приводящие к анемии;
· консультация оториноларинголога – носовые кровотечения;
· консультация онколога – злокачественное поражение, которое является причиной кровотечения;
· консультация нефролога – исключение заболеваний почек при дифференциальной диагностике с анемией хронических заболеваний;
· консультация фтизиатра – кровотечение или вторичная анемия на фоне туберкулеза;
· консультация пульмонолога – кровопотери или вторичная анемия на фоне заболеваний бронхолегочной системы;
· консультация гинеколога – кровотечение из половых путей (ювенильные кровотечения, дисменореи и т.д.);
· консультация эндокринолога – снижение функции щитовидной железы, наличие диабетической нефропатии;
· консультация гематолога – для исключения заболеваний системы крови и при неэффективности ферротерапии у пациентов с верифицированным дефицитом железа;
· консультация проктолога – ректальные кровотечения;
· консультация инфекциониста – при наличии признаков гельминтоза или другого паразитоза, приводящего к анемии.
Диагностический алгоритм: см. Приложение 1
Дифференциальный диагноз и обоснование дополнительных исследований:
Железодефицитная анемия до получения результатов исследования обмена железа на основании ОАК требует проведения дифференциальной диагностики с другими гипохромными анемиями, вызванными нарушением синтеза гемоглобина. К ним относятся анемии, связанные с нарушением синтеза порфиринов (анемия при свинцовом отравлении, при врожденных нарушениях синтеза порфиринов), а также талассемии. Гипохромные анемии в отличие от железодефицитных анемий протекают с высоким содержанием железа в крови и депо, которое не используется для образования гема (сидероахрезия), при этих заболеваниях отсутствуют признаки тканевого дефицита железа. Дифференциальным признаком анемии, обусловленной нарушением синтеза порфиринов, является гипохромная анемия с базофильной пунктацией эритроцитов, ретикулоцитов, усиленным эритропоэзом в костном мозге с большим количеством сидеробластов. Для талассемии характерны мишеневидная форма и базофильная пунктация эритроцитов, ретикулоцитоз и наличие признаков повышенного гемолиза.
Верификация дефицита железа и ЖДА проводится только на основании лабораторных данных – снижения уровня гемоглобина, ферритина, сывороточного железа, TSat и др. в связи с чем дифференциальная диагностика с нозологиями, при которых не характерен дефицит железа (миелодиспластический синдром, апластическая анемия, витамин В12 дефицитная анемия, фолиеводефицитная анемия, гемолитические анемии) не вызывает затруднений.
В случаях, когда у пациента с анемией и лабораторно подтвержденным дефицитом железа со стороны системы крови выявляются симптомы или лабораторные изменения, которые не характерны для ЖДА, требуется дообследование у гематолога.
Дифференциальная диагностика при лабораторно подтвержденной ЖДА требуется с другими заболеваниями и состояниями при которых возможно развитие сидеропении [1].
| Диагноз | Обоснование для дифференциальной диагностики | Обследования | Основные критерии диагноза |
| Латентный дефицит железа | Характерно снижение уровня сывороточного железа | Сывороточное железо, мкмоль/л | ê |
| TSat, % | ≥16% | ||
| Ферритин, мкг/л | |||
| Гемоглобин, г/л | Норма | ||
| MCV [2] | ê или в норме | ||
| MCH [3] | ê или в норме | ||
| Дополнительно: | |||
| sTFR, мг/л | é | ||
| Содержание гемоглобина в ретикулоцитах, пг | |||
| Функциональный дефицит железа | Возможно снижение уровня сывороточного железа | Сывороточное железо, мкмоль/л | ê или в норме |
| TSat, % | ê или в норме | ||
| Ферритин, мкг/л | Норма | ||
| Гемоглобин, г/л | Норма | ||
| MCV | Норма | ||
| MCH | Норма | ||
| Дополнительно: | |||
| sTFR, мг/л | é | ||
| Содержание гемоглобина в ретикулоцитах, пг | |||
| Функциональный дефицит железа | Возможно снижение уровня сывороточного железа | Сывороточное железо, мкмоль/л | ê или в норме |
| TSat, % | ê или в норме | ||
| Ферритин, мкг/л | Норма | ||
| Гемоглобин, г/л | Норма | ||
| MCV | Норма | ||
| MCH | Норма | ||
| Дополнительно: | |||
| sTFR, мг/л | é | ||
| Содержание гемоглобина в ретикулоцитах, пг | |||
| IRIDA (ведущий признак – неэффективность ферротерапии) | Характерно снижение уровня сывороточного железа и TSat | Сывороточное железо, мкмоль/л | ê |
| TSat, % | |||
| Ферритин, мкг/л | вариабельно | ||
| Гемоглобин, г/л | ê | ||
| MCV | êê | ||
| MCH | êê | ||
| Дополнительно: | |||
| sTFR, мг/л | é | ||
| Содержание гемоглобина в ретикулоцитах, пг | ê | ||
| Анемия хронических заболеваний | Характерно снижение сывороточного железа | Сывороточное железо, мкмоль/л | ê |
| TSat, % | ê или в норме | ||
| Ферритин, мкг/л | >100 | ||
| Гемоглобин, г/л | ê | ||
| MCV | ê или в норме | ||
| MCH | ê или в норме | ||
| Дополнительно: | |||
| sTFR, мг/л | ê или в норме | ||
| Содержание гемоглобина в ретикулоцитах, пг | ê | ||
| ЖДА и анемия хронических заблеваний | Характерно снижение сывороточного железа | Сывороточное железо, мкмоль/л | ê |
| TSat, % | ê или в норме | ||
| Ферритин, мкг/л | |||
| Гемоглобин, г/л | ê | ||
| MCV | êê | ||
| MCH | êê | ||
| Дополнительно: | |||
| sTFR, мг/л | вариабельно | ||
| Содержание гемоглобина в ретикулоцитах, пг | ê | ||
| ЖДА | Характерно снижение сывороточного железа, TSat и ферритина | Сывороточное железо, мкмоль/л | ê |
| TSat, % | |||
| Ферритин, мкг/л | |||
| Гемоглобин, г/л | ê | ||
| MCV | ê | ||
| MCH | ê | ||
| Дополнительно: | |||
| sTFR, мг/л | é | ||
| Содержание гемоглобина в ретикулоцитах, пг | ê |
Получить консультацию по медтуризму

Получить консультацию по медтуризму
| Аскорбиновая кислота (Ascorbic acid) |
| Железа (III) гидроксид декстран (Ferric (III) hydroxide destrane) |
| Железа (III) гидроксид полимальтозат (Ferric (III) hydroxide polymaltosate) |
| Железа (III) гидроксид сахарозный комплекс (Ferric (III) hydroxide sacharose complex) |
| Железа глюконат (Ferrous gluconate) |
| Железа карбоксимальтозат (Ferric carboxymaltosate) |
| Железа сульфат (Ferric sulfate) |
| Железа фумарат (Ferrous fumarate) |
| Фолиевая кислота (Folic acid) |
ТАКТИКА ЛЕЧЕНИЯ НА АМБУЛАТОРНОМ УРОВНЕ [6-16]
ЖДА является хроническим заболеванием, развитие которого требует времени, за которое успевают включиться механизмы адаптации и в большинстве случаев достигается компенсация. Пациенты сохраняют неплохое самочувствие и клинические проявления ЖДА минимальны. Поэтому лечение ЖДА должно проводиться амбулаторно. При использовании парентеральных препаратов железа возможна госпитализация в дневной стационар.
Лечебная программа при ЖДА включает:
· устранение этиологических факторов (лечение основного заболевания);
· лечебное питание (диета № 11);
· лечение железосодержащими препаратами;
· восполнение запасов железа (терапию насыщения).
· противорецидивную терапию.
Немедикаментозное лечение:
· Диета. При железодефицитной анемии больному показана диета, богатая железом. Железо из продуктов животного происхождения всасывается в кишечнике в значительно больших количествах, чем из растительных продуктов.
Медикаментозное лечение:
ЖДА не может быть успешно излечена в случае, если не устранена её причина. Основой патогенетической терапии ЖДА являются препараты железа. Трансфузии не заменяют ферротерапию. Ферротерапия может проводиться препаратами железа для приема внутрь и парентеральными препаратами.
Из парентеральных препаратов предпочтительнее использование внутривенных, т.к. введение внутримышечных болезненно, имеет вариабельную абсорбцию и может приводить к формированию инфильтратов.
Конечный результат терапии препаратами железа вне зависимости от пути введения одинаков – рост уровня гемоглобина. Отличия между различными препаратами с различными путями введения заключаются в переносимости лечения и темпах прироста уровня гемоглобина.
Препараты железа для приема внутрь
Основными принципами лечения ПЖ для приема внутрь являются следующие:
· назначение ПЖ с достаточным содержанием элементарного железа;
· нецелесообразность одновременного назначения витаминов группы В (в том числе В12), фолиевой кислоты без специальных показаний в связи с отсутствием доказательств преимуществ в эффективности и безопасности перед монокомпонентными препаратами, невозможностью отследить фармакокинетику препарата при наличии 3 и более компонентов в 1 таблетке;
· избегание назначения препаратов железа внутрь при наличии признаков нарушения всасывания в кишечнике;
· достаточная продолжительность насыщающего курса терапии (не менее 3 мес., может увеличиваться до 5-6 месяцев);
· необходимость проведения поддерживающей терапии ПЖ после нормализации показателей гемоглобина в соответствующих ситуациях.
Рекомендуемая суточная доза элементарного железа у большинства взрослых составляет 150-200 мг. [9] Применение более высоких доз не имеет смысла, поскольку всасывание железа при этом не увеличивается. Например, таблетка железа сульфата массой 325 мг содержит 65 элементарного железа, три таблетки – 195 мг железа из которых может быть адсорбированно и утилизированно только 25 мг. [10]
Терапия препаратами железа для приема внутрь должна проводиться в течение 3х месяцев для восполнения запасов в депо. [9]
При пероральной ферротерапии наиболее распространены гастроинтестинальные побочные эффекты. Они включают металлический вкус, тошноту, диарею, запор, потемнение стула.
Для уменьшения выраженности побочных эффектов возможно уменьшение дозы (перевод на однократный прием, например) или увеличение интервала приёма, переход на прием другого препарата железа с меньшим содержанием элементарного железа, переход с таблеток на жидкие лекарственные формы, которые позволяют более просто подбирать переносимую дозу, отмена пероральных препаратов и назначение внутривенных.
Причины неэффективности терапии ПЖ для приема внутрь:
· отсутствие дефицита железа (неправильная трактовка природы гипохромной анемии и ошибочное назначение ПЖ);
· недостаточная дозировка ПЖ (недоучет количества трехвалентного железа в препарате);
· недостаточная длительность лечения ПЖ;
· нарушение всасывания ПЖ, назначаемых внутрь больным с соответствующей патологией;
· одновременный прием препаратов, нарушающих всасывание железа;
· наличие хронических (оккультных) кровопотерь, чаще всего из органов ЖКТ;
· сочетание ЖДА с другими анемическими синдромами (В12-дефицитной, фолиеводефицитной).
Парентеральные препараты железа.
Показания для назначения парентеральных препаратов железа [1, 11]:
· нарушение всасывания при патологии кишечника (энтериты, синдром недостаточности всасывания, резекция тонкого кишечника, резекция желудка по методу Бильрот II с выключением двенадцатиперстной кишки);
· выраженные гастроинтестинальные побочные эффекты пероральной терапии, не устранимые другими способами;
· постоянные потери крови, при которых потребность в железе превышает физиологические возможности для всасывания железа (например, тяжелое маточное кровотечение, наследственная геморрагическая телеангиоэктазия с поражением слизистых оболочек);
· пожелание пациента о быстром (за 1-2 визита) восполнении дефицита железа и отказ от продолжительной многомесячной ферротерапии;
Кумулятивную дозу, необходимую для восстановления уровня гемоглобина в крови и восполнения запасов железа в организме, при внутривенном введении ПЖ вычисляют по формуле Ганзони:
*депо железа у человека с массой тела >35 кг и = 35 кг = 500 мг и
| Лекарственная группа (МНН) | Лекарственные средства | Способ применения | Уровень доказательности | |
| Монокомпонентные препараты железа B03A (код АТХ) | ||||
| Глюконат железа** | Глюконат железа* 300 мг | Пероральный прием по 2 таб. х 2-3 раза в день | III C | |
| Сульфат железа** B03AA07 | Сульфат железа табл. 256.3 мг (80 мг железа), Сульфат железа 325 мг (105 мг иона (II) железа (Fe 2+ )) | Пероральный прием по 1таб. х 2 раза в день | III C | |
| Фумарат железа** B03AA02 | Фумарат железа* (суспензия 3 гр) для детей, Железа фумарат 200 – таб. 200 мг (65 мг железа), Железа фумарат капс.300 мг (100 мг эл. Железа) | Пероральный прием: железа фумарат по 1таб. х 3 раза в день, по 1 капс. х 2-4 раза в день | III C | |
| Комбинированные препараты В03АА | ||||
| В03АА Железа (II) сульфат сухой + Аскорбиновая кислота 60 мг | Сульфат железа таб.320 мг+аскорбиновая кислота 60 мг | Пероральный прием, по 1 таб. х 1-2 раза в сутки | III C | |
| Фумарат железа, фолиевая кислота B03AD02 | Железа фумарат 163,56 мг (50 мг железа) и фолиевая кислота 540 мкг | Перорально по 1 капс. х 2 раза в сутки | III C | |
| Препараты железа (III) валентные | ||||
| Железа (III) гидроксид полимальтозат** | Железа (III) гидроксид полимальтозат 400 мг (100 мг эл.железа) Таб. 375 мг (100 мг эл.железа) | Пероральный прием по 1 жеват. таб. х 2-3 р в день | IIB | |
| Препараты железа для парентерального введения. | ||||
| Лекарственная группа (МНН) | Лекарственные средства | Способ применения | Уровень доказательности | |
| B03AC02 Железа оксида сахарат | Железо III гидроксид сахарозный комплекс 540 мг (железа 20 мг); | |||
Железа (III) гидроксида сахарозного комплекса 333,0 мг (эквивалентно элементарному железу) 20,0 мг
В/в медленно (0,2 мл/мин) в дозе 100–200 мг железа (2–4 мл), предпочтительно разведенного в 10–20 мл 0,9% раствора натрия хлорида или 5% раствора глюкозы. Начальная доза препарата составляет 25 мг железа или 0,5 мл раствора, которая вводится в/в медленно в течение 1–2 мин
в/в (внутривенно) капельно (инфузионно) 1000 мг железа 1 раз в неделю.
*применение препарата после регистрации на территории РК
**нет в КНФ
Хирургическое вмешательство:
Проводятся при продолжающихся кровотечениях на стационарном уровне.
Дальнейшее ведение:
· Для пациентов, получающих пероральную ферротерапию контрольные исследования ОАК следует проводить через 1-2 недели от начала лечения.
· При терапии парентеральными препаратами, особенно с однократным введением, контрольные исследования ОАК могут проводится через 4-8 недель от введения препарата.
· Пациенты с продолжающимися кровотечениями (например, с наследственной геморрагической телеангиоэктазией) нуждаются в более частом контроле.
NB! Причинами рецидива ЖДА могут быть недостаточная длительность приема пероральных препаратов, продолжающиеся потери крови, некорректный диагноз ЖДА, наличие дополнительных причин для развития анемии.
Индикаторы эффективности лечения: см. стационарный уровень.
ТАКТИКА ЛЕЧЕНИЯ НА СТАЦИОНАРНОМ УРОВНЕ [7-9]
Тактика лечения аналогична амбулаторному уровню. При гемодинамической нестабильности может проводиться трансфузия эритроцитсодержащих компонентов крови.
Эритроцитсодержащие компоненты крови при ЖДА следует переливать только в случаях гемодинамической нестабильности (УД – С; см. ниже) и подобная практика не должна быть рутинной [7].
Критерии гемодинамической нестабильности (должны присутствовать все признаки) [8]:
· Систолическое артериальное давление менее 100 мм.рт.ст.;
· Изменения сознания;
· Одышка/тахипное;
Количество трансфузий нужно максимально ограничивать. Уровень гемоглобина не является определяющим для оценки показаний к трансфузиям и выбору тактики лечения, т.к. пациенты могут иметь разную степень адаптации к анемии и клинические проявления являются более значимыми.
· Перечень основных лекарственных средств (имеющих 100% вероятность применения) – см. Амбулаторный уровень;
· Перечень дополнительных лекарственных средств (менее 100% вероятности применения) – см. Амбулаторный уровень.
Хирургическое вмешательство:
Показаниями к хирургическому лечению является продолжающееся кровотечение, нарастание анемии, вследствие причин, которые не могут быть устранены путем медикаментозной терапии.
Дальнейшее ведение: см. Амбулаторный уровень.
Индикаторы эффективности лечения [9]:
· разрешение симптомов;
· ретикулоцитарный криз на 7-10 дни от начала терапии препаратами железа, может быть не выражен при анемии легкой степени;
· повышение уровня гемоглобина происходит как правило медленно, начиная с 1-2 недель лечения с последующим приростом примерно 20 г/л в течение последующих трех недель с нормализацией к 6-8 неделям от начала лечения;
· контролировать показатели обмена железа рекомендуется не ранее, чем 4 недели после завершения терапии. Терапия препаратами железа завершается, если показатели обмена железа (TSat и ферритин) нормализуются. В случае дискордантности между TSat и ферритином рекомендуется ориентироваться на первый показатель.
Госпитализация в круглосуточный стационар может проводиться только в исключительных случаях при наличии признаков гемодинамической нестабильности, обусловленной только подтверждённой ЖДА при исключении всех других причин гемодинамической нестабильности.
Показания для плановой госпитализации: нет.
Показания для экстренной госпитализации:
Экстренная госпитализация пациента с ЖДА показана только при наличии признаков гемодинамической нестабильности при исключении других причин гемодинамической нестабильности:
· при выявленном источнике кровотечения в зависимости от источника кровопотери в отделение хирургического профиля (хирургию, гинекологию, проктологию и т.д.);
· при отсутствии установленного кровотечения – госпитализация в терапевтическое отделение.
- Протоколы заседаний Объединенной комиссии по качеству медицинских услуг МЗ РК, 2017
- 1) Camaschella C. Iron-Deficiency Anemia /N Engl J Med 2015; 372:1832-1843. 2) Peyrin-Biroulet L.,Williet N., Cacoub P. Guidelines on the diagnosis and treatment of iron deficiency across indications: a systematic review /Am J Clin Nutr doi: 10.3945/ajcn.114.103366. 3) Сельчук В.Ю. Чистяков С.С. Толокнов Б.О. и соавт. Железодефицитная анемия: современное состояние проблемы /РМЖ, 2012. №1:1. 4) Sarzynski E. I., Puttarajappa C., Xie Y., Grover M., Laird-Fick H. Association between proton pump inhibitor use and anemia: a retrospective cohort study // Dig Dis Sci. 2011, Aug; 56 (8): 2349–2353. 5) Dado DN, Loesch EB, Jaganathan SP A Case of Severe Iron Deficiency Anemia Associated with Long-Term Proton Pump Inhibitor Use Curr Ther Res Clin Exp. 2017 Jan 21;84:1-3. 6) Auerbach M, Ballard H, Glaspy J. Clinical update: intravenous iron for anaemia. Lancet 2007; 369:1502. 7) Red Blood Cell Transfusion: 2016 Clinical Practice Guidelines from the AABB (Journal of the American Medical Association; October 12, 2016. 8) San Luis Obispo County EMS Agency ALS Treatment Protocols 2007 [Rev. 11/1/10] 9) Schrier S., Auerbach M. Treatment of iron deficiency anemia in adults / www.uptodate.com, aug, 2017. 10) Schrier SL. So you know how to treat iron deficiency anemia. Blood 2015; 126:1971. 11) Auerbach M, Deloughery T. Single-dose intravenous iron for iron deficiency: a new paradigm. Hematology Am Soc Hematol Educ Program. 2016 Dec;2016(1):57-66. 12) Rodgers GM, Auerbach M, Cella D, Chertow GM, Coyne DW, Glaspy JA, Henry DH High-molecular weight iron dextran: a wolf in sheep’s clothing? J Am Soc Nephrol. 2008;19(5):833 13) Administration of intravenous iron sucrose as a 2-minute push to CKD patients: a prospective evaluation of 2,297 injections.Macdougall IC, Roche A/Am J Kidney Dis. 2005;46(2):283. 14) Safety and efficacy of rapidly administered (one hour) one gram of low molecular weight iron dextran (INFeD) for the treatment of iron deficient anemia. Auerbach M, Pappadakis JA, Bahrain H, Auerbach SA, Ballard H, Dahl NV/Am J Hematol. 2011 Oct;86(10):860-2. Epub 2011 Aug 29. 15) Intravenous iron treatment in pregnancy: comparison of high-dose ferric carboxymaltose vs. iron sucrose. Christoph P, Schuller C, Studer H, Irion O, De Tejada BM, Surbek D/J Perinat Med. 2012;40(5):469. Epub 2012 May 13. 16) Intravenous ferric carboxymaltose compared with oral iron in the treatment of postpartum anemia: a randomized controlled trial. Van Wyck DB, Martens MG, Seid MH, Baker JB, Mangione A/Obstet Gynecol. 2007;110(2 Pt 1):267.
ОРГАНИЗАЦИОННЫЕ АСПЕКТЫ ПРОТОКОЛА
Список разработчиков протокола с указание квалификационных данных:
1) Пивоварова Ирина Алексеевна – председатель РОО «Казахстанское Общество врачей-гематологов» Республики Казахстан, Председатель Правления ТОО «Центр гематологии».
2) Загурская Елена Юрьевна – заместитель Председателя Правления ТОО «Центр гематологии».
3) Клодзинский Антон Анатольевич – кандидат медицинских наук, гематолог ТОО «Центр гематологии», медицинский советник, заместитель председателя РОО «Казахстанское Общество врачей – гематологов» Республики Казахстан.
4) Хан Олег Ромуальдович – ассистент кафедры внутренних болезней, гематолог РГП на ПХВ «Научно- исследовательский институт кардиологии и внутренних болезней».
5) Юхневич Екатерина Александровна – и.о. доцента кафедры клинической фармакологии и доказательной медицины РГП на ПХВ «Карагандинский государственный медицинский университет», клинический фармаколог
Указание на отсутствие конфликта интересов: нет.
Рецензенты:
Тургунова Людмила Геннадиевна – доктор медицинских наук, профессор, заведующая кафедрой терапевтических дисциплин факультета непрерывного профессионального развития, врач-гематолог РГП на ПХВ «Карагандинский государственный медицинский университет».
Указание условий пересмотра протокола: пересмотр протокола через 5 лет после его опубликования и с даты его вступления в действие или при наличии новых методов с уровнем доказательности.
Приложение 1
источник
АНЕМИЯ /anaemia, греч. an- и haima — бескровие, синоним малокровие/ — состояние, характеризующееся уменьшением числа эритроцитов и снижением уровня гемоглобина в единице объема крови.
По классификации Г.А.Алексеева /1970/, анемии делят на три основные группы: вследствие кровопотерь — постгеморрагические; вследствие нарушенного кровообразования; вследствие повышенного кроверазрушения — гемолитические.
Как указывает А.И. Воробьев /1985/, классифицирование анемий на основе патогенеза и назологическое классифицирование анемий невозможно с позиций научной строгости, поскольку каждую анемию нельзя вместить в рамки группы, к которой она принадлежит. Например, постгеморрагическая анемия железодефицитная относится к группе анемий, связанный с кровопотерей, и к группе анемий с нарушениями образования крови. К анемиям, вызванным нарушением кровообразования в детском возрасте, относят железодефицитную, витамин-В12-фолиево-дефицитную и апластические анемии.
За рубежом в основе классификации анемий лежит средний эритроцитарный объем /СЭО/ эритроцитов. Соответственно выделяют микро-, макро- и нормоцитарные анемии. Форма эритроцитов — серповидноклеточная, анемия, мишеневидная и т.д.
К МИКРОЦИТАРНЫМ АНЕМИЯМ относятся: железодефицитная анемия, малая и большая b-талассемия, отравление свинцом, анемии при хронических заболеваниях.
МАКРОЦИТАРНЫЕ АНЕМИИ часто сопровождаются мегалобластозом костного мозга, поэтому и классифицируются по наличию или отсутствию мегалобластоза.
МЕГАЛОБЛАСТНАЯ АНЕМИЯ наблюдается при дефиците витамина В12 и фолиевой кислоты, применении лекарственных средств, нарушающих метаболизм фолиевой кислоты /метотрекстат, триметоприм/, болезнях обмена веществ /оротовая ацидурия/, выделяют также тиамин-зависимую анемию.
ИЗОЛИРОВАННАЯ МАКРОЦИТАРНАЯ АНЕМИЯ /без мегабластоза/ встречается при заболеваниях печени, поражении костного мозга, гипотиреозе, применении некоторых лекарственных средств /вальпроевая кислота/.
НОРМОЦИТАРНЫЕ АНЕМИИ чаще возникают при кровотечениях, гемолизе и снижении продукции эритроцитов.
При КРОВОТЕЧЕНИИ необходимо выявить источник кровотечения. Острое кровотечение сопровождается пропорциональной потерей плазмы и клеток, поэтому уровни Нв и Нt не изменяются. Анемия возникает только после восстановления ОЦК /примерно через 3 суток/. Ретикулоцитоз возникает через 6-12 часов после кровопотери в связи с мобилизацией запасов костного мозга; развернутая гемолитическая картина наблюдается через 5-10 сут. Хроническая кровопотеря приводит к дефициту железа и, как следствие, к микроцитозу и уменьшению числа ретикулоцитов.
ГЕМОЛИТИЧЕСКИЕ АНЕМИИ развиваются в результате преждевременного разрушения эритроцитов и включают множество заболеваний.
ПРИЧИНАМИ гемолиза эритроцитов могут быть: механическое повреждение эритроцитов /искусственные клапаны сердца, травма/, термическое повреждение, микроангиопатии, гиперспленизм, ауто- и изоиммунные анемии, отравления грибами, болезнь Вильсона /повышается уровень меди в крови/, инфекционные заболевания, нарушение структуры мембраны эритроцитов, гемоглобина, ферментопатии. Отсюда все гемолитические анемии делятся по принципу уточнения фактора, вызвавшего гемолиз.
Наследственные анемии делятся на следующие группы:
С нарушением мембраны эритроцитов — наследственный микросфероцитоз, наследственный овалоцитоз.
Связанные с нарушением активности ферментов эритроцитов — дефицит глюкозо — 6 — фосфат — дегидрогиназы.
Связанные с нарушением структуры и синтеза гемоглабина — В — талассемия и серповидноклеточная анемия.
Из наследственных гемолитических анемий у детей чаще встречается микросфероцитоз, реже бывает дефицит Г-6-ДГ. В тропических странах распространены семейные талассемия и серповидноклеточная анемия. Из приобретенных анемий чаще встречается — гемолитическая болезнь новорожденных, в тропиках — анемии, обусловленные дефицитом витаминов, паразитарные.
ДИАГНОСТИКА. В пользу гемолитической анемии свидетельствует бледность кожных покровов с желтушным оттенком, иногда внезапно возникшие. При лабораторном обследовании отмечается повышение уровня непрямого билирубина в сыворотке, снижение сывороточного гаптоглобина, при тяжелом гемолизе — снижение гемоглобина и гемоглобинурия. Отмечается изменение формы эритроцитов: микросфероциты, мишеневидные и т.д. Дальнейшее обследование включает проведение пробы Кумбса, электрофорез Нв, определение осмотической резистентности. Осмотическая резистентность при гемолитических анемиях резко снижается. Обязательно исследуется геммограмма с подсчетом числа ретикулоцитов. В зависимости от степени гемолиза отмечается рост числа ретикулоцитов. Во время гемолитических кризисов число их может достигнуть 100%. Число тромбоцитов всегда в норме. Число лейкоцитов в период кризисов растет, с нейтрофильным сдвигом в лево. В зависимости от уровня Цветного показателя /ЦП/ анемии могут быть нормохромные, гипохромные, гиперхромные. Все гемолитические анемии — нормо — или гиперхромные, за исключением талассемии, которая всегда гипохромная. Отмечается снижение числа эритроцитов и уровня Нв, а также изменение эритроцитометрической кривой Прайс-Джонса. Если средний диаметр нормальных эритроцитов 7,2-7,5 мкм, то при гемолитических анемиях кривая Прайс-Джонса растянута за счет анизацитоза, поскольку имеются эритроциты с диаметром от 4 до 9 мкм и более. Реакция Кумбса, как правило отрицательная. Костный мозг очень богат клеточными элементами за счет гиперплазии красного ростка.
ТАЛАССЕМИЯ /мишеневидно-клеточная анемия, болезнь Кули/ характеризуется повреждением /делецией/ гена, ответственного за синтез a-или b-цепей, При a- талассемии нарушен синтез a-цепей. Поскольку данная цепь входит в состав всех нормальных фракций гемоглобина, при a-талассемии наблюдается равномерное снижение этих цепей. В основе b-талассемии лежит наследственное угнетение b-цепей, входящих в состав НвА. Описаны случаи g-, d-, bd-талассемии с нарушением синтеза соответствующих цепей глобина. Чаще встречается b-талассемия.
Талассемия распространена в основном в зоне субтропического климата, в Средиземноморье, среди населения Юго-Восточной Азии, Африки, на островах Тихого океаны. Спорадические случаи встречаются во всех районах земного шара, они являются спорадически возникшими мутациями. Поскольку гемозиготы погибают, не достигнув детородного возраста, то отбор идет в пользу гетерозигот.
Малая талассемия (носительство признака (-талассемии).
Гетерозилотная форма (-талассемии сопровождается незначительной анемией. Уровень гемоглобина в среднем на 20-30% ниже возрастной нормы. Эритрциты гипохромны, отличаются микроцитарностью, отмечается пойкило- и овалоцитоз и часто грубые базофильные включения, Встречаются мишеневидные клетки, но в небольшом количестве; их не следует рассматривать в качестве специфического для таласемии признака, Может умеренно сокращаться продолжительность жизни эритроцитов, однако явные признаки гемолиза, как правило отсутствуют. Уровень железа в сыворотке не изменен или повышен.
У носителей гена талассемии нередко ошибочно ставят диагноз железодефцитной анемии, по поводу чего их в течение длительного времени лечат препаратами железа. Более чем у 90% лиц, носителей гена, уровень НвА2 повышен до 3,4-7,0%, что имеет диагностическое значение. Почти у половины из них несколько повышен (в пределах 2-6%) уровень НвF.
Если мать или отец являются носителями гена, при каждой беременности риск развития большой талассемии составляет 25%. Методики, позволяющие произвести забор крови плода, создают возможность ее препатальной диагностики. С помощью прямой аспирации крови из вен плаценты при фетоскопии, выполняемой в 16-20 недель беременности, может быть полчена небольшая проба крови плода. Ее инкубируют с лейцином 14С, после чего можно количественно оценить синтез (-,(- и (-цепей. У гомозиготных по (-талассемии плодов с помощью этого метода удается продемонстрировать заметное снижение синтеза (-цепей.
Выявление пораженных плодов сможет быть осуществлено при использовании ферментов, ограничивающих эндонуклеазу, для анализа ДНК фибробластов, полученных из амнистической жидкости. Биопсия трофобластов позволяет установть дианоз у плода в возрасте10-12 недель.
Большая В-талассемия /гомозиготная форма/ протекает тяжело вследствие гемолиза и нарушенного эритропоэза. Известна как болезнь Кули.
Патогенез. При талассемии не синтезируется или мало синтезируется одна из цепей глобина. В норме синтез цепей сбалансирован и свободных цепей нет. При талассемии появляются избыточные цепи глобина, что является главной причиной неэффективного эритропоэза. Эритрокариоциты разрушаются в костном мозге, а эритроциты и ретикулоциты периферической крови — в селезенке. При b-талассамии происходит избыточное накопление в эритроцитах фетального гемоглобина /НвF/, который обладает повышенным сродством к кислороду, в результате растет тканевая гипоксия, нарушается рост и развитие ребенка. Избыток миелокариоцитов в скелете вызывает деформацию костей.
КЛИНИЧЕСКАЯ картина. Проявления большой талассемии начинаются на первом году жизни. Характерна бледность, тяжелая анемия — гемоглобин до 60-20 г/л, эритроциты до 2 млн. В мазках периферической крови-мишеневидные эритроциты анизо- и пойкилоцитоз, гипохромия эритроцитов.
Увеличение числа клеток эритроидного ростка в костном мозге, является не истинной гиперплазией красного ростка, а результатом накопления функционально неполноценных эритроидных элементов. Увеличение их содержания происходит за счет накопления ядросодержащих клеток красного ростка, не способных к дифферецировке, т.е. отмечается неэффективный эритропоэз.
Кроме анемии и желтухи, для талассемии характерны отставание в росте и изменения в костной ткани /особенно со стороны костей черепа/, деформация черепа, приводящая к формированию “лица больного анемией Кули” — башенный череп, увеличение верхней челюсти, монголоидный разрез глаз, выступление резцов и клыков с нарушением прикуса. Рентгенологически определяется симптомом “волосатого черепа” или “ежика”, так называемый игольчатый периостоз. В длинных трубчатых костях расширены костномозговые полости, кортикальный слой истончен, часты патологические переломы.
Ранний признак талассемии — значительное увеличение печени и селезенки. При развитии гиперспленизма на фоне- лейко- и тромбоцитопении развиваются вторичные геморрации, инфекционные осложнения. Начиная с 8-10 лет возникают осложнения, связанные с перенасыщением органов железом / гемосидерозом внутренних органов/. Гемосидероз миокарда с развитием сердечной недостаточности — основная причина смерти больных анемией Кули.
Гепатомегалия является проявлением гемосидероза и заканчивается фиброзом, формированием цирроза печени. Фиброз поджелудочной железы осложняется развитием сахарного диабета; может развиться калькулезный холецистит. Снижение концентрационной способности почек усугубляет гипоксическую дистрофию. Даже больные, прошедшие лечение, умирают в возрасте 17 лет. Однако при хорошем диспансерном наблюдении и периодическом введении десферала срок жизни больного может быть продлен.
Выделяют 3 степени тяжести гомозиготной В-талассамии:
Тяжелую, развившуюся с первых месяцев жизни ребенка и быстро заканчивающуюся смертью ребенка;
Хроническую, наиболее часто встречающуюся, при которой дети доживают до 5-8 лет;
Легкую при которой больные доживают до взрослого возраста.
Выделяют также малую /гетерозиготную форму/ В-талассамии. Наблюдается у практически здоровых лиц со сбалансированным синтезом глобинов в костном мозге и отсутствием неэффективного эритропоэза, однако у них обнаруживаются мишеневидные эритроциты.
ЛЕЧЕНИЕ. Малоэффективно. Оно состоит в основном из трансфузионной терапии отмытыми эритроцитами и введения хелатора десферала (суточная доза 250-500 мг.), способствующего экскреции с мочой значительного количества железа, особенно при насыщении организма аскорбиновой кислотой. Назначают препараты, необходимые при напряженном эритропоэзе, в частности фолиевую кислоту и витамин В12, другие витамины группы В /В1,6,15/ и витамин Е. Применение глюкокортикоидов считается неоправданным.
При выраженной спленомегалии показана спленэктомия. По возможности операцию откладывают до 5 летнего возраста, чтобы уменьшить риск сепсиса.
СЕРПОВИДНОКЛЕТОЧНАЯ АНЕМИЯ — также относится к гемоглабинопатиям — широко распространена в странах Средиземноморья, Ближнего и Среднего Востока, Индии, Америке. Серповидноклеточная анемия /СКА/ — гемоглобиноз S, “реологическая болезнь” описана в начале 20 в., когда были открыты эритроциты серповидной формы, в 1956 году было доказано, что Нв S отличается от Нв А тем, что в положении 6 в цепи глютаминовая кислота заменена на валин.
ЭТИОЛОГИЯ. Предполагают, что этот метаморфоз произошел впервые при инвазии плазмодия малярии как защитный акт, а затем стал наследоваться. Наследуется заболевание аутосомно и носит кодоминантный характер, поскольку нарушения выявляются у обоих партнеров, отсюда высокий уровень заболеваемости и смертности, обусловленный гомозиготностью.
ПАТОГЕНЕЗ. Замещение глютамина валином приводит к тому, что у Нв S вместо отрицательного заряда, характерного для Нв А, появляется нейтральный, а это усиливает связь одной молекулы с другой молекулой гемоглобина. Внутри эритроцита Нв переходит в состояние геля, а при пониженном парциальном давлении кислорода осаждается в виде тактоидов — веретенообразных остроконечных кристаллов. Тактоиды растягивают эритроциты придавая им серповидную форму. Появление серповидных эритроцитов значительно повышает вязкость крови, что уменьшает скорость кровотока и приводит к закупорке мелких капилляров. S-эритроциты теряют пластичность, застревают в капиллярах с последующим тромбозом /окклюзией/ сосудов, возникают инфаркты, сопровождаемые гипоксией.
КЛИНИЧЕСКАЯ КАРТИНА. При физикальном обследовании больных выявляются бледность кожи и слизистых оболочек, желтушность, усиливающаяся с возрастом. У младших детей начиная с 6 месяцев пальпируется селезенка, однако к 8 годам спленомегалию обнаруживают редко. Это объясняется фиброзом селезенки, развившимся на фоне частых инфарктов. У 60% детей обнаруживают гепатомегалию. Нередко отмечают кардиомегалию, при этом над областью сердца выслушивают различной интенсивности шумы. Характерным симптомом является аденопатия, в желчном пузыре выявляются камни /холелитиаз/, часто встречается язва 12 перстной кишки.
Больные СКА имеют характерный вид: удлиненный нижний сегмент тела, дорсальный кифоз и люмбальный лордоз, куполообразное /“готическое”/ небо, выступающий лоб и “башенный” череп, значительное удлинение конечностей. Нарушение процессов роста у этих детей имеет определенные закономерности: в возрасте до 3 лет, как правило, нет отставания от сверстников, затем некоторая задержка до 7-8 лет, а в взрослом возрасте больные имеют нормальный рост и даже несколько выше. Дети отстают в половом развитии, однако уровень интелектуального развития как правило нормальный.
Заболевание протекает хронически, больные тяжелой формой живут около 20 лет Бывают периодически острые состояния — кризы, которые делятся на 2 группы: 1 -клинические /болевые или вазоокклюзионные/, при которых показатели состава гемоглобина и ретикулоцитов не отличаются от нормы; 2- гематологические с резким снижением уровня Нв и высоким ретикулоцитозом. Нередко кризы сочетаются.
КЛИНИЧЕСКИЕ КРИЗЫ /болевые, вазоокклюзионные, секвестрационные / начинаются на 2-3 году жизни ребенка. Они провоцируются инфекционными заболеваниями, лихорадкой, дегидратацией организма, физическим переутомлением. Болевой синдром связан с возникновением инфарктов вследствие окклюзии серповидными эритроцитами сосудов. Инфаркты могут быть в костном мозге, костях, перартикулярных тканях суставов. Основной признак окклюзии — боль различной интенсивности с повышением температуры, возможным симметричным опуханием тыльных поверхностей кистей стоп – серповидно-клеточный дактилит,синдром “рук-ног”,что может быть первым клиническим проявлением. Рентгенологически наблюдаются остеосклеротические изменения. В костном мозге возникают некрозы, инфаркты, с повышением температуры, беспокойства, расстройства сознания. Со стороны легких — отдышка, кровохарканье. Со стороны ССС- тахикардия, цианоз.
Секвестрационный криз происходит при захвате серповидных эритроцитов селезеночными синусами, которые являются местом гибели утративших свою силу S –эритроцитов, что приводит к депонированию крови в селезенке и печени, уменьшению ОЦК , развитию гиповолемического шока. Причем быстрое увеличение селезенки сопровождается побледнением кожи и слизистых оболочек, напряжением мышц живота из-за болезненности селезенки, симптомами сердечной недостаточности, рвотой. У детей со спланомегалией наблюдаются частые рецидивы, и это нередко приводит к смертельному исходу.
ГЕМАТОЛОГИЧЕСКИЕ КРИЗЫ. /апластический, гипергемолитический, мегалобластный /
Наиболее тяжелый — апластический криз наблюдается у детей 5-8 лет. В результате временного прекращения образования эритроцитов резко падает уровень гемоглобина, исчезают ретикулоциты. Наступает аплазия только красного ростка; нарушение лейко- и тромбоцитопоэза нет. Отмечается бледность кожных покровов слизистых оболочек, лихорадка, боли. Боли в костях, почти всегда имеются признаки сердечной недостаточности. При этом в крови содержится около 20 г/л Нв.
Гипергемолитический криз развивается в результате резкого гемолиза эритроцитов. Кроме бледности и лихорадки, для этого криза характерно нарастание желтушности.
Мегалобластический криз имеет много общего с апластическим, но в данном случае кроме резкого снижения гемоглобина и ретикулоцитов, в костном мозге происходит мегабластная гиперплазия красного ростка. При этом кризе большое значение придают хроническому дефициту фолиевой кислоты.
ЛЕЧЕНИЕ. Специальных методов лечения серповидноклеточной анемии не существует, применяется симптоматическая терапия.
Назначаются анальгетики и гидратационная терапия с целью уменьшения вязкости крови. Назначают ингибиторы образования серповидных эритроцитов, в частности препараты цинка, повышающие химическое сродство гемоглабина к кислороду. /цинк уменьшает число серповидных эритроцитов, благодаря воздействию на мембранную проницаемость/. Такой же эффект обнаружен у новокаина. Показан эритроцитафарез с последующим введением эритроцитов. Трансфузии эритромассы рекомендуются при тяжелой степени анемии (НВ менее 60-70 г/л) и апластических кризах (4 мл эритроцитарной массы повышают уровень гемоглобина на 1 г/л.).
Учитывая тяжесть течения и неблагоприятный прогноз при серповидноклеточной анемии и талассемии необходимо проведение медико-генетического консультирования в семьях.
Пренатальная диагностика позволяет установить диагноз у плода с 10-12 нед.(при биопсии трофобласта), с 16-20 недель возможно проведение аспирации из вен плаценты.
Наследственный сфероцитоз — НС /болезнь Минковского-Шоффара/ является широко распространенным заболеванием (2-3 случая на 10000 населения) и встречается у лиц большинства этнических групп, однако чаще болеют жители северной части Европы.
ЭТИОПАТОГЕНЕЗ. Заболевание передается по аутосомно-доминантному типу. Как правило, у одного из родителей выявляют признаки гемолитической анемии. Возможны спорадические случаи заболевания (в 25%), представляющие собой новые мутации. В патогенезе НС бесспорны 2 положения: наличие генетически детерминированной аномалии белков, или спектринов, мембраны эритроцитов и элиминирующая роль селезенки в отношении сфероидальноизмененных клеток. У всех больных с НС отмечен дефицит спектринов в эритроцитарной мембране /до 1/3 нормы/, а у некоторых — нарушение их функциональных свойств, причем установлено, что степень дефицита спектрина может коррелировать с тяжестью заболевания.
Наследственный дефект структуры мембраны эритроцитов приводит к повышенной проницаемости ее для ионов натрия и накоплению воды, что в свою очередь ведет к чрезмерной метаболической нагрузке на клетку, потере поверхностных субстанций и формированию сфероцита. Формирующиеся сфероциты при движении через селезенку начинают испытывать механическое затруднение, задерживаясь в красной пульпе и подвергаясь всем видам неблагоприятных воздействий (гемоконцентрация, изменение рН, активная фагоцитарная система), т.е. селезенка активно наносит сфероцитам повреждения, вызывая еще большую фрагментацию мембраны и сферуляцию. Это подтверждается при электронно-микроскопических исследованиях, позволивших обнаружить ультраструктурные изменения в эритроцитах /утолщение клеточной мембраны с ее разрывами и образованием вакуолей/. Через 2-3 пассажа через селезенку сфероцит подвергается лизису и фагоцитозу. Селезенка является местом гибели эритроцитов; продолжительность жизни которых сокращается до 2 недель.
Хотя дефекты эритроцитов при НС обусловлены генетически, в организме возникают условия при которых углубляются эти дефекты и реализуется гемолитический криз. Кризы могут провоцироваться инфекциями, некоторыми химическими веществами, психическими травмами.
КЛИНИКА. Заболевание может проявляться с неонатального периода, однако более выраженные симптомы обнаруживают к концу дошкольного и в начале школьного возраста. Раннее проявление заболевания предопределяет более тяжелое течение. Чаще болеют мальчики.
НС — гемолитическая анемия с преимущественно внутриклеточным типом гемолиза, это обусловливает и клинические проявления болезни — желтуху, увеличение селезенки, большую или меньшую степень анемии, склонность к образованию камней в желчном пузыре.
Жалобы, клинико-лабораторная симптоматика во многом определяются периодом заболевания. Вне гемолитического криза жалобы могут отсутствовать. При развитии гемолитического криза отмечаются жалобы на повышенную утомляемость, вялость, головную боль, головокружение, бледность, желтуху, снижение аппетита, боли в животе, возможны повышение температуры до высоких цифр, тошнота, рвота, учащение стула, грозный симптом — появление судорог.
Симптоматика криза во многом определяется анемией и зависит от степени гемолиза.
При объективном обследовании кожа и видимые слизистые бледные или лимонно-желтые. У детей с ранними проявлениями НС возможны деформации скелета, особенно черепа (башенный, квадратный череп, изменяется расположение зубов и т.д.); нередки генетические стигмы. У больных обнаруживаются разной степени выраженности изменения со стороны сердечно-сосудистой системы, обусловленные анемией. Характерен гепатолиенальный синдром с преимущественным увеличением селезенки. Селезенка плотная, гладкая, нередко болезненная, что, по-видимому, объясняется напряжением капсулы вследствие кровенаполнения или периспленитом. Окраска экскрементов в момент криза интенсивная. Следует отметить возможные колебания в размерах селезенки: значительное увеличение при гемолитических кризах и уменьшение в период относительного благополучия.
В зависимости от тяжести НС клинические симптомы могут быть выражены незначительно. Иногда желтуха может быть единственным симптомом, по поводу которой больной обращается к врачу. Именно к этим лицам относится известное выражение Шоффара: “Они более желтушны, чем больны.”. Наряду с типичными классическими признаками заболевания встречаются формы НС, когда ГА может быть настолько хорошо компенсирована, что пациент узнает о заболевании лишь при проведении соответствующего обследования.
Наряду с наиболее типичными гемолитическими кризами при тяжелом НС возможны арегенераторные кризы с симптомами гипоплазии преимущественно красного ростка костного мозга. Такие кризы могут развиваться остро с довольно яркими симптомами анемии-гипоксии и наблюдаются обычно у детей после 3 лет жизни. Арегенераторные кризы кратковременны (1-2 недели) и носят обратимый характер в отличие от истинной аплазии.
НС осложняется образованием пигментных камней в желчном пузыре и желчных протоках, после 10 лет камни желчного пузыря встречаются у половины больных, не подвергнутых спленэктомии.
Диагноз НС ставится на основании генеалогического анамнеза, клинических данных, описанных выше и лабораторных исследований. Гемолитическую природу анемии подтверждают нормохромная нормоцитарная анемия с ретикулоцитозом, непрямая гипербилирубиннемия, степень выраженности которых зависит от тяжести гемолиза. Окончательный диагноз основывается на морфологических особенностях эритроцитов и характерном признаке НС — изменении осмотической резистентности эритроцитов. К морфологическим особенностям эритроцитов при НС относятся шарообразная форма (сфероциты), уменьшение диаметра (сред. диаметр э.
Снижение осматич. резистентности
Признак выражен: постоянно +, непостоянно ± , отсутствует-.
ДЕФИЦИТ АТИВНОСТИ Г-6-ФД ЭРИТРОЦИТОВ
Дефицит активности глюкозо-6-фосфатдегидрогеназы (Г-6-ФД) встречается наиболее часто в государствах, расположенных на побережье Средиземного моря (Италия, Греция), в Африке и Латинской Америке и др. странах
Этиология. Причину нарушения активности Г-6-ДФ эритроцитов у населения различных этнических групп связывают с распространением малярии. Было обнаружено, что в нормальных эритроцитах паразитов меньше, чем в эритроцитах с дефицитом Г-6-ФД. Хорошо известна наследственная передача этого заболевания с Х-хромосомой.
Патогенез. В эритроцитах со сниженной активностью фермента Г-6-ФД уменьшается образование восстановленного некатинамидаденин-динуклеотидфосфата (НАДФ) и связывание кислорода, а также снижается скорость восстановления метгемоглобина и понижается устойчивость к воздействию различных потенциальных окислителей – аскорбиновой кислоты, метиленового синего и др. Известно более 40 медикаментов (хинин, сульфаниламидные препараты, нитрофураны, левомицетин, амфотерицин, аспирин и др.), не считая вакцин и вирусов, ряд растительных продуктов, которые потенциально способны вызывать острый внутрисосудистый гемолиз в эритроцитах у лиц с дефицитом Г-6-ФД.
Клиническая картина. Недостаточность Г-6- ФД отличается приемущественно у лиц мужского поля, обладающих, как известно, единственной Х-хромосомой. У женщин клиника гомозиготности на 3-5-й день после приема терапевтической дозы того или иного препарата.
Выделяют пять клинических форм недостаточности Г-6-ФД в эритроцитах: 1) острый внутрисосудистый гемолиз – классическая форма недостаточности Г-6-ФД. Чаще встречается у представителей европеоидной и монголоидной рас. Развивается в результате приема лекарств, вакцинации, диабетического ацидоза, в связи с вирусной инфекцией; 2) фавизм, связанный с употреблен6ием в пищу или вдыханием цветочной пыльцы некоторых бобовых (Vicia fava); 3) гемолитическая болезнь новорожденных, не связанная с гемоглобинопатией, с групповой или резус-несовместимостью, осложняющегося иногда “ядерной желтухой”; 4) наследственная хроническая гемолитическая анемия (несфероцитарная), обусловленная недостаточностью Г-6-ФД в эритроцитах; 5) бессимптомная форма.
Клинически заболевание проявляется желтушностью, увеличением печени,
В периферической крови выражена анемия с ретикулоцитозом, лейкоцитозом со сдвигом до миелоцитов. Отмечается анизо-, пойкилоцитоз, видны осколки эритроцитов (шизоциты), полихромазия, базофильная пунктация эритроцитов.
Характерный признак внутрисосудистого гемолиза – гипергемоглобинемия, сыворотка крови при стоянии приобретает коричневый цвет за счет образующегося метгемоглобина. Одновременно отмечается гипербилирубинемия, моча может быть цвета черного пива, что обусловлено выделяющимися гемоглобином, метгемоглобином, а также гемосидерином и уробилином.
Лечение. Начинается с отмены лекарства, вызвавшего гемолитический криз. При нетяжелом кризе назначаются антиоксиданты (эревит, vit Е). Применяются средства, способствующие увеличению восстановленного глутатиона в эритроцитах, количество которого снижается при гемолитических кризах, ксилит по 0,25-0,5 г. 3 раза внутрь. Одновременно дается фенобарбитал (или зиксорин) в суточной дозе в зависимости от возраста детям по 0,005-0,01 в течение 10 дней. Фенобарбитал, обладает билирубинконъюгирующим действием, индуцирует глюкуронилтрансферазную систему печени.
При тяжелых гемолитических кризах для профилактики острой почечной недостаточности внутривенно капельно в зависимости от возраста вводят 1-4% раствора натрия гидрокарбоната из расчета 5 мл на 1 кг массы в сутки, что способствует выведению продуктов гемолиза. Как слабый диуретик и антиагрегант тромбоцитов, улучшающий почечный кровоток, применяется 2,4% р-р эуфиллина внутривенно капельно 4-6 мг/кг в сутки в 250-500 мл физ. раствора.
ОСТРЫЙ ЛИМФОБЛАСТНЫЙ ЛЕЙКОЗ
Лейкозы – являются первичным опухолевым заболеванием костного мозга, при котором опухолевые клетки, поражая костный мозг, распространяются не только по органам кроветворения, но и в ЦНС, и в другие органы и системы. Это наиболее частые злокачественные новообразования у детей (частота 1:20000), в возрасте до 4 лет.
Согласно современной схеме кровотворения, острые лейкозы объединяет общий признак: субстракт опухоли составляют бластные клетки. При хронических лейкозах субстрактом опухоли являются созревающие и зрелые клетки.
По имеющимся данным, лейкозы “полиэтиологичны ”, т.к. не установлено какой-либо безусловно вызывающей лейкоз причины. Причинными факторами лейкоза человека могут быть химические (экзо- и эндогенные) и физические (ионизирующая радиация) факторы, а также вирусы.
Рауншенбах М.О. с соавт., 1974г., обнаружил у больных лейкозом людей некоторые в-ва из метаболитов триптофана и тирозина, которые способны индуцировать лейкозы и опухоли у мышей.
У человека найден вирус лимфомы Беркитта и выявлена транскриптаза, которая способствует синтезу ДНК на вирусной РНК, что приводит к образованию эндосимбиоза онкогенного вируса и клетки. Это позволило считать обоснованной вирусную этиологию лейкозов.
По гипотезе Р. Хабнера, 1976 г., в геноме каждой клетки заложена информация в виде ДНК-провируса, равноценная информации в геноме онковируса. В норме ДНК-провирус (онкоген) находится в репрессированном состоянии, однако под воздействием концерогенных факторов (химических, радиации) он активизируется и вызывает клеточную трансформацию. Провирус передается по наследству. Некоторые ученые допускают возможность существования систем, подавляющих вирусную лейкозную трансформацию в клетках хозяина, в частности системы, ответственной за иммунитет. Таким образом, в этиологии заболевания главную роль играет не инфицированность вирусом, а состояние контролирующих систем, стимулирующих факторов.
Радиационный этиологический фактор в последние десятилетия привлек внимание как реально существующий фактор внешней среды и внутренней среды человека, способный вызывать лейкоз.
Радиациональные лейкозы человека реально существуют. Это лейкозы у жителей Японии, возникающие много лет спустя после взрывов атомных бомб в Хиросиме и Нагосаки.
Нами была выявлена связь между ростом заболеваемостью лейкозом и загрязнением воздуха некоторыми полютантами воздуха в таких районах Крыма как Сакский, Бахчисарайский, Раздольненский и др.
Итак, этиология лейкозов находится в стадии изучения.
О патологической сущности лейкозов известно, что лейкоз это опухоль. В основе его лежит первичная патология клеток кроветворения, сопровождающаяся нарушением процессов их пролиферации и дифференцирования, возникновение клонов опухолевых (лейкозных) клеток. Прогрессирующая прлиферация этих клеток приводит к поражению органов кроветворения и уменьшению плацдарма нормального гемопоэза.
Учение о развитии лейкозного процесса, понятие об опухолевой прогрессии при лейкозах ввел А.И. Воробъев в 1965 г. В настоящее время это учение разработано применительно ко всем формам лейкоза с соответствующими патогенетическими мероприятиями терапии.
Бластные клетки при остром лейкозе теряют ферментную специфичность. Клетки становятся морфологически и цитохимически недифференцируемыми. Они характеризуются:
Изменением ядра и цитоплазмы (вместо крупных появляются клетки неправильной формы с увеличением площади ядра и цитоплазмы;
Обладают способностью расти вне органов гемопоэза (пролифераты из лейкозных клеток находят в коже, почках, головном мозге и в мозговых оболочках), они неравноценны и представляют разные этапы прогрессии;
Имеют скачкообразный уход опухоли из-под цитостатического воздействия, а также лучевого, гормонального;
Нарастание процесса в виде выхода бластных элементов в периферическую кровь, перехода от лейкопении к лейкоцитозу.
Этапы опухолевой прогрессии – это этапы злокачественности лейкоза. В основе данной прогрессии лежит нестабильность генетического аппарата лейкозных клеток, которым свойственен переход из неактивного состояния в активное. Раскрытие этапов лейкозного процесса имеет большое практическое значение, ибо главный его смысл заключается в поисках цитостатических препаратов, адекватных каждому этапу злокачественности.
Начиная разбор клинической картины лейкозов, необходимо коснуться вопроса о современной классификации заболевания, стадиях и диагностике.
Начиная с 1976 г. во всем мире клиницисты пользуются ФАБ (Франция, Америка, Британия) классификацией острых лейкозов. По которой лейкозы делятся на острый лимфобластный лейкоз /ALL/, 75-80% от всех лейкозов у детей, и острый нелимфобластный лейкоз ANLL, который включает в себя семь подтипов (М1 – М7) и составляет 20-25% лейкозов у детей.
М1,2 – острый миелобластный лейкоз, М 1 – недифференцируемая острая миелоидная лейкемия, М 2 – дифференцируемая миелоидная лейкемия, 15-18%, чаще у детей старшего возраста.
М 3 – острый промиелоцитарный лейкоз, 2-5%.
М 4 – острый миелобластный лейкоз, встречается очень редко.
М 5 – острая монобластная и моноцитарная лейкемия.
М 6 – острый эритромиелоз (эритролейкоз) – 1,25%.
М 7 — острая мегакариоцитарная лейкемия.
М 0 – острый недифференцируемый лейкоз.
Выживаемость при острых нелимфобластных лейкозах значительно ниже, чем при ОЛЛ. Однако в настоящее время она значительно возросла в связи с трансплантацией костного мозга от братьев или сестер, совместимых по HLA.
Для установления диагноза лейкоза необходимо цитологическое, цитохимическое и цитогенетическое исследование костного мозга и выявление поверхностных антигенных маркеров клеток (иммунофенотипирование L1, L2, L3).
С учетом морфологической характеристики ОЛЛ выделяют 3 подварианта (см. таблицу 4).
Сравнительная цитологическая характеристика острого лимфобластного и острого миелобластного лейкоза у детей.
ALL – L 1, малая, однороная,гомогенная популяция.
Скудная с редкими вакуолями
0-1, от мелких до неопределенных.
ALL – L 2 от средних до больших; разнородная, гетерогенная популяция.
от умеренной до большой; непостояннобазофильная с редкими вакуолями.
от круглых до неправильных, уродливых.
1, различимые или неразличимые.
ALL – L 3 от средних до больших.
AМL – М 1 от средних до больших.
от средней до большой, палочки Ауэра
от круглых до овальных, хроматин от тонкого до грубого.
1 или больше, обычно различимые.
ПРИМЕЧАНИЕ: М 1 – острая миелобластная лейкемия без зрелых форм.
Цитохимические реакции, характерные для острых лейкозов.
PAS – периодическая Шифф-кислота (реакция на гликоген)
ANB- альфа нафтил бутиратэстераза
Как видно из таблицы 5 L 1 и L 2 варианты ОЛЛ в большинстве PAS положительные, а L 3 вариант – отрицательный, в то время как нелимфобластные лейкозы дают положительную реакцию на миелопероксидазу и судан черный.
Для клинической практики выделение трех подвариантов ОЛЛ имеет очень большое значение, поскольку от подварианта зависит курс проводимой терапии и прогноз для жизни больного. Кроме того L 1 и L 2 подварианты могут быть Т-клеточной формой или ни-Т- ни В-клеточной формой, а L 3 подвариант В-клеточной формой.
При постановке диагноза ОЛЛ необходимо указать и стадию заболевания. В 1979 г. А.И. Воробьев и М.Д. Бриллиант предложили свою классификацию ОЛЛ. Согласно этой классификации выделяют начальный период, развернутую стадию болезни, полную ремиссию, выздоровление (состояние полной ремиссии на протяжении 5 лет), частичную ремиссию, рецидив с указанием, какой по счету, и уточнением очага локализации при локальном рецидиве, терминальная стадия.
Для прогноза заболевания имеет значение диагностирование на ранней стадии. Ни у кого нет сомнения сегодня, что ОЛЛ начинается постепенно и лишь его манифестация представляется острой. Многие исследователи пишут о “предлейкозе ”, но в предлейкозе поставить диагноз нельзя. Поставить диагноз лейкоза позволяет морфологическая картина костного мозга с наличием 30% бластов и более. В начальном периоде ОЛЛ протекает под видом болезней, “масок ”, связанных преимущественно с гиперпластическим поражением органов, либо с выраженным цитопеническим синдромом. Соответственно детям выставляется диагноз ревматизм, лимфаденит, инфекционный мононуклеоз, злокачественная лимфома и др., а с другой стороны апластическая анемия, геморрагический васкулит, тромбоцитопеническая пурпура, сепсис и др.
Следует подчеркнуть, что для раннего распознавания ОЛЛ у детей во всех неясных случаях о лейкозе надо думать чаще и прибегать к исследованию костного мозга. Особенно это имеет значение для постановки диагноза в алейкемическую стадию, когда в геммограмме еще отсутствуют бласты. К сожалению диагноз ОЛЛ чаще выставляется в лейкемическую стадию, при наличии бластов в геммограмме.
Пунктат костного мозга особенно необходимо исследовать в случаях, сопровождающихся цитопенией или увеличением лимфоузлов, либо тем и другим вместе, не применяя до этого никакого лечения (рентгеновское облучение, электрофорез, УВЧ, назначение преднизалона), так как такая терапия может ускорить течение лейкозного процесса и миелограмма будет нехарактерной, что затруднит постановку диагноза, оставит больного на долгое время без специфической терапии.
Для манифестных клинических проявлений ОЛЛ характерны увеличение лимфатических узлов, печени, селезенки, боли в костях и суставах, повышение температуры, бледность, быстрая утомляемость, недомогание, геморрагии. Реже появляется стоматит, головная боль, обмороки, изменения со стороны нервной системы.
В периферической крови находят либо алейкемическую картину, либо наличие бластных клеток, в костном мозге – 30% и более бластов. Если же диагноз острого лейкоза неубедителен (10-15% бластных клеток), то, возможно только симптоматическое лечение, необходимо повторно исследовать миелограмму через 3-4 недели.
В стадии полной ремиссии в пунктате костного мозга находят не более 5% бластных клеток, а общее количество бластных и лимфоидных клеток в нем не превышает 40%; в периферической крови бластных клеток не должно быть, в составе крови возможны умеренная лейкемия и тромбоцитопения, соответственно 3,0 * 10 9 /л и 100,0 * 10 9 /л из-за цитотоксического воздействия; должен быть нормальный состав спиномозговой жидкости и отсутствовать клинические признаки лейкозной пролиферации.
Частичная ремиссия может характеризоваться гематологическим улучшением, уменьшением бластных клеток в костном мозге, в спиномозговой жидкости при ликвидации клинических признаков нейролейкоза и (или) при подавлении очагов лейкозной инфильтрации в других органах, вне костного мозга.
Рецидив острого лейкоза бывает костномозговым (появление в пунктате более 5% бластных клеток) или внекостномозговым с различной локализацией лейкозной инфильтрации (нейролейкоз, лейкозная инфильтрация селезенки, лимфоузлов, гайморовых пазух, яичек и др.).
В терминальную стадию, клинически в значительной мере условную, вкладывается морфологическое понятие об исчерпании терапевтических резервов, о некурабельном этапе опухолевой прогрессии лейкоза. Вместе с этим отмечается условность этой стадии, поскольку она отражает лишь современный уровень терапевтических возможностей. Все цитостатические средства не только оказываются неэффективными, но и на их фоне прогрессирует лейкоз, нарастает цитопенический синдром, прежде всего агранулоцитоз, появляются некрозы на слизистых оболочках, сепсис, кровоизлияние. Терминальная стадия свидетельствует о необходимости отмены цитостатической терапии с сохранением симптоматической.
Причины смерти детей при лейкозах, в том числе лимфобластном, – это сепсис, кровоизлияния в мозг, интоксикация.
При остром лимфобластном лейкозе (ОЛЛ) выделяют прогностически неблагоприятные формы, которые влияют на выживаемость больного.
Особо выделяется нейролейкоз, который может присоединиться к любым локальным формам рецидива и стать самостоятельной формой, без проявления какой-либо другой клиники заболевания. В последние годы нейролейкоз стал частым проявлением рецидива заболевания. Это связывают с увеличением продолжительности жизни больных и с тем обстоятельством, что противолейкозные препараты не проникают через гематоэнцефалический барьер и для лейкозных клеток в центральной нервной системе создается определенное укрытие, благоприятные условия для проявления лейкозных инфильтратов.
НЕЙРОЛЕЙКОЗ (лейкозное поражение нервной системы) развивается вследствие метастазирования лейкознык клеток в оболочки головного и спинного мозга, в вещество мозга и нервные стволы. Диагноз нейролейкоза ставится на основании обнаружения бластных клеток в ликворе. При нейролейкозе в ликворе может наблюдаться вариобельный цитоз до нескольких тысяч клеток в 1 мкл., повышение уровня белка и снижение уровня глюкозы, цвет жидкости, как правило, сероватый, с положительной реакцией Панди.
Клинических проявлений при нейролейкозе, кроме обнаружения цитоза в ликворе, может и не наблюдаться; в то же время они могут быть самыми разнообразными. Это изменение поведения ребенка: раздражительность, вялость, необщительность. Появляется головная боль, тошнота, рвота (при менингиальной форме), очаговая симптоматика в виде поражения черепных нервов, пирамидной недостаточности, мозжечковых симптомов, судорог, нарушения речи и сознания (при менингоэнцефалической и энцефалической форме ), нижних парапарезов, нарушение походки и функции тазовых органов, корешковых симптомов (при менингомиелитической форме). Экзофтальм наблюдается, как правило, при хлорлейкозе.
Почти у половины больных при рецидиве лейкоза отмечается нормальный состав периферической крови, и поэтому большое значение имеет внимательный осмотр больного, своевременная костно-мозговая и люмбальная пункции при диспансерном наблюдении.
Прогностические признаки при ОЛЛ у детей (по Л.А. Махоновой и др., 1986г.).
Период от начала болезни до постановки диагноза.
Увеличение периферических лимфоузлов.
Морфология бластов по ФАБ-классификации.
Цитохимические данные: кислая фосфотаза.
Клинические маски острого лейкоза самые разнообразные: лимфаденит, эпид, паротит, туберкулез, лимфогранулематоз, ревматизм, ревматоидный артрит, гепатиты, инфекционный мононуклеоз, острый аппендицит, затяжные простудные заболеванияы, длительные язвено-некротические стоматиты, ангины, сепсис, апластическая анемия, гемолитическая анемия, тромбоцитопеническая пурпура, геморрагический васкулит, дизентерия, мелкоочаговая пневмония.
Тромбоцитопения – ниже 100*10 9
ЛЕЧЕНИЕ ОСТРОГО ЛИМФОБЛАСТНОГО ЛЕЙКОЗА.
С 1990 года в гематологическом отделении Республиканской клинической детской больницы применяют современные терапевтические протоколы лечения острого лимфобластного лейкоза у детей ОЛЛ-БФМ-90 разработанные немецкими коллегами (1988, 1990, 1992гг.). По протоколам среди детей, больных ОЛЛ, выделяют три группы риска:
Группа стандартного риска – дети от 1 года до 6 лет. Количество бластов на 8 день терапии в периферической крови не превышает 1000 в 1 мкл. (после 7-дневного приема преднизалона); отсутствует пре-Т иммунофенотип лейкоза (если у пациента не проводилось иммуноспецифическое исследование бластов, но имеется медиастенальная опухоль, то пациент в любом случае из стандартной группы риска); не имеется первичного поражения ЦНС; установлена полная ремиссия на 33 –й день лечения;
Группа среднего риска – дети до 1 года и старше 6 лет, количество бластов в периферической крови на 8 –й день, после 7-дневной преднизолоновой профазы, не превышает 1000 в 1 мкл; полная ремиссия на 33 –й день лечения;
Группа высокого риска – отсутствие полной ремиссии на 33 –й день лечений.
Между 65 и 70 днем повторная контрольная костномозговая пункция для подтверждения ремиссии, если на 33 день в костном мозге 5% и более бластов
(в гемограмме не более 20 х 10 9 /л лейкоцитов).
КЛАССИФИКАЦИЯ ЦИТОСТАТИЧЕСКИХ СРЕДСТВ.
1 Антиметаболиты – нарушают синтез предшественников нуклеиновых кислот, путем конкуренции с последними в лейкозной клетке.
Метотрексат – антогонист фолиевой кислоты (эффект в стадии разгара и в качестве поддерживающей терапии, действует на S фазу).
Ланвис (Tioguaninum) ( Glaxo Wellcome) — 1 табл. содержит 40 мг тиогуанина : 25 табл. в упаковке. Тиогуанин является сульфгидрильным аналогом гуанина и проявляет свойства пуринового антиметаболита. Являясь структурными аналогами пуриновых нуклеотидов, метаболиты тиогуонина включаются в пуриновый обмен и ингибируют синтез нуклеиновых кислот в опухолевых клетках.
6-Меркаптопурин (пуринетол) – антогонист пурина, вмешивается в обмен нуклеиновых кислот, действует на S фазу.
Цитозар (цитозинарабинозид) – систематический аналог пиримидина, препятствует превращению цитидина в диоксицитидин (на S фазу лейк. клетки).
2 Алкилирующие соединения – подавляют синтез ДНК и в меньшей степени РНК в лейкозной клетке.
циклофосфан, обладающий цитостатическим и цитолитическим действием на лейкозные клетки.
Винкристин – практически воздействует на все фазы покоящейся клетки.
Этопозид (вепезид, VP –16) – из растений мондрагоры, предотвращает вхождение клетки в митоз.
L-аспарагиназа (краснитин) – разлагает аспарагин, необходимый для синтеза протеина, на аспарагиновую кислоту и амоний в лейкозной клетке, которая не способна к самостоятельному синтезу аспарагина и поэтому погибает от его эндогенного действия.
5 Противоопухолевые антибиотики.
Адриамицин, рубомицин – обладает цитостатическим действием, подавляет синтез нуклеиновых кислот путем взаимодействия с ДНК и РНК – полимеразами.
Преднизолон – обладает цитолитическим действием на лейкозные клетки (лимфобласты и недифференцируемые бласты) и не вызывает разрушения нормальных лимфоцитов.
Дозировка медикаментов осуществляется в соответствии с площадью поверхности тела пациента. Каждый раз перед началом нового этапа лечения вновь определяется площадь поверхности тела и расчитывается соответственно доза препаратов. При эндолюмбальной инъекции и при облучении головы дозировка осуществляется в соответствии с возрастом.
Полихимиотерапия больных со стандартным и средним факторами риска длится в течение 6-ти месяцев.
ПРОТОКОЛ I (направлен на редукцию ремиссии), см. рис.4.

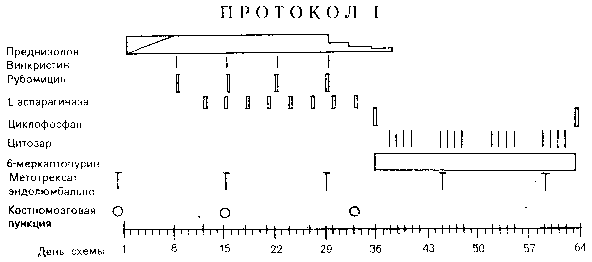 Первая фаза. Преднизолон назначается из расчета 60 мг/м 2 с 1-го по 28 день, затем снижение дозы на 50% каждые три дня до полной отмены. У пациента с высокой начальной массой лейкозных клеток или выраженной органомегалией начальную дозу необходимо назначать из расчета 20 мг/м 2 , до снижения уровня лейкоцитов в периферической крови ниже 20 * 10 9 /л, чтобы избежать острого синдрома распада белков.
Первая фаза. Преднизолон назначается из расчета 60 мг/м 2 с 1-го по 28 день, затем снижение дозы на 50% каждые три дня до полной отмены. У пациента с высокой начальной массой лейкозных клеток или выраженной органомегалией начальную дозу необходимо назначать из расчета 20 мг/м 2 , до снижения уровня лейкоцитов в периферической крови ниже 20 * 10 9 /л, чтобы избежать острого синдрома распада белков.
В зависимости от клинического ответа на терапию (уменьшение органов), лабораторных показателей (уменьшение числа лейкоцитов, содержания мочевой кислоты, мочевины, креатинина, калия) дозу преднизалона нужно быстро увеличивать до окончательной (60 мг/м 2 ) не позднее 5-го дня терапии.
Винкристин вводится в дозе 1,5 мг/м 2 внутривенно на 8, 15, 22, 29-й день.
Рубомицин вводится из расчета 30 мг/м 2 внутривенно капельно в течении 1 часа на 8, 15, 22, 29-й день. Перед первым введением рубомицина делается ЭКГ и эхография сердца (препарат кардиотоксичен).
L-Аспариназа (L-ASP) – вводится в дозе 10000 ед/м 2 внутривенно капельно в течении 1 часа на 8, 15, 18, 21, 24, 30, 33-й день.
Возможны тяжелые аллергические реакции, гипергликемия и нарушение свертывания крови. Необходимо проводить лабораторный контроль свертывания крови (фибриноген, АТ-Ш, плазминоген), уровня сахара. При развитии анафилаксии возможна замена на эрвиназу.
Профилактика нейролейкоза проводится эндолюмбальными введениями метотрексата в возрастной дозировке 1 раз в 2 недели.
Вторая фаза. Условия для начала фазы 2 протокола 1 следующие: полная ремиссия в костном мозге, менее 5% бластов в миелограмме; при начальном поражении ЦНС – полная ремиссия в ЦНС; при медиастенальной опухоли – уменьшение размеров опухоли до 30% ее начальной величины; удовлетворительное общее состояние; отсутствие тяжелой инфекции; креатинин в пределах возрастной нормы; лейкоциты более 2 * 10 9 /л; гранулоциты – более 0,5 * 10 9 /л.
Циклофосфан (ЦФ) – вводится в дозе 1000 мг/м 2 внутривенно капельно в течении часа на 36-й и 64-й день лечения. Контроль диуреза и профилактика геморрагического цистита проводятся следующим образом: гидратация 3000 мл/м 2 за 24 часа; фуросемид из расчета 0,5 мг/кг внутривенно на 6 и 12 час после циклофосфана; месна (урометоксан) 400мг/м 2 внутривенно на 0,4 и 8 часу от начала инфузии циклофосфана.
6-Меркаптопурин (6-МП) — 60 мг/м 2 в сутки внутрь, 36- 63-й дни лечения.
Цитозар (ARA-C) — 75 мг/м 2 в сутки внутривенно в виде 4 дневных блоков; 38-41 дни, 45-48, 52-55, 59-62 дни. После начала лечения цитозаровые блоки желательно не прерывать. Если начало цитозаровых блоков откладывается или прерывается, одновременно прекращается прием 6-МП. Выпавшее в лечении 6-МП нужно подтянуть до общей плановой кумулятивной дозы, равной 1680 мг/м 2 .
ПРОТОКОЛ М ( направлен на консолидацию ремиссии), см. рис.5.

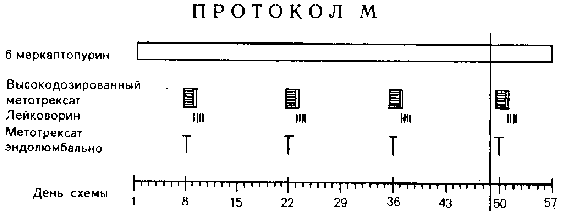 включает в себя 6-меркаптопурин, высокодозированный метотрексат, метотрексат эндолюмбально, лейковорин внутривенно. Начинается через 2 недели после завершения протокола 1. Основные условия для начала этого протокола: лейкоциты больше 1,5 * 10 9 /л; гранулоциты больше 0,5 * 10 9 /л; тромбоциты больше 50 * 10 9 /л.
включает в себя 6-меркаптопурин, высокодозированный метотрексат, метотрексат эндолюмбально, лейковорин внутривенно. Начинается через 2 недели после завершения протокола 1. Основные условия для начала этого протокола: лейкоциты больше 1,5 * 10 9 /л; гранулоциты больше 0,5 * 10 9 /л; тромбоциты больше 50 * 10 9 /л.
6-Меркаптопурин назначается из расчета 25 мг/м 2 в день (внутрь), в течение 8 недель.
Метотрексат назначается в дозе 1 г/м 2 ( 1/10 общей дозы, вводится в течение 30 мин; 9/10 общей дозы вводится в течение 35,5 ч.) путем непрерывной инфузии, на 8, 22, 36, 50 день от начала протокола М.
Лейковорин (антидот метотрексата) дается из расчета 15 мг/м 2 внутривенно струйно или внутрь в таблетках на 42, 48, 54 час от начала введения.
МТХ интралюмбально вводится через 2 часа от начала внутривенной инфузии МТХ в возрастной дозировке.
ПРОТОКОЛ II (также направлен на консолидацию ремиссии), см. рис.6.

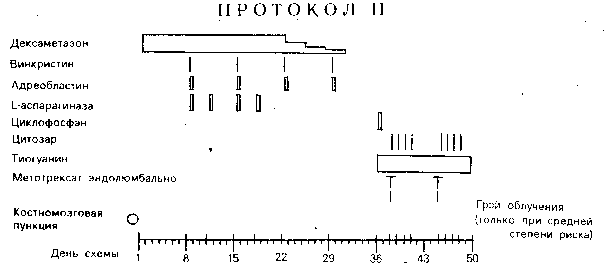 Первая фаза. Проводится через две недели после протокола М. Условия начала П протокола следующие: продолжается полная ремиссия; хорошее общее состояние больного; отсутствие инфекции; лейкоциты больше 2,5* 10 9 /л; гранулоциты больше 1 * 10 9 /л; тромбоциты больше 100 * 10 9 /л.
Первая фаза. Проводится через две недели после протокола М. Условия начала П протокола следующие: продолжается полная ремиссия; хорошее общее состояние больного; отсутствие инфекции; лейкоциты больше 2,5* 10 9 /л; гранулоциты больше 1 * 10 9 /л; тромбоциты больше 100 * 10 9 /л.
Дексаметазон 10 мг/м 2 внутрь с 1 по 21 день от начала протокола, затем дозу уменьшают каждые 3 дня на 50% до полной отмены.
Винкристин 1,5 мг/м 2 – 4 внутривенных введения с интервалом в одну неделю (максимальная доза 2 мг/м 2 ).
Адриамицин (ADR) 30 мг/м 2 , инфузия в течение 1 часа. Перед 1 и 3 назначением необходимо провести ЭКГ и эхокардиографическое исследование: при признаках снижения сократительной функции миокарда следует прекратить дальнейшее применение ADR.
L-Аспарагиназа 10000 ЕД/м 2 – инфузия в течение 1 часа на 8, 11, 15, 18 день.
Вторая фаза. Условия для начала второй фазы протокола П: хорошее общее состояние; отсутствие инфекций; нормальный возрастной уровень креатинина в сыворотке крови; лейкоциты больше 2 * 10 9 /л; гранулоциты больше 0,5 * 10 9 ; тромбоциты больше 50 * 10 9 /л.
Циклофосфан 100 мг/м 2 , инфузия в течение 1 часа на 36 день от начала протокола. Контроль диуреза и профилактика цистита такие же, как в протоколе 1.
6-Тиогуанин (6-TG) 60 мг/м 2 внутрь в 36-49 дни, всего 14 дней.
Цитозар (ARA-C) 75 мг/м 2 внутривенно двумя блоками через 4 дня: 38-41 день, 45-48 дни.
МТХ интралюмбально вводится в возрастных дозировках на 38 и 45 день от начала протокола.
Облучение головы. При хорошем клиническом состоянии облучение головы начинается с 38 дня протокола П для детей из групп среднего и высокого риска. Доза облучения зависит от возраста и от начального вовлечения ЦНС (от 12 до 18 Гр детям до 2 лет, от 18 до 24 Гр детям старше 2 лет).
В случае выявления нейролейкоза люмбальные пункции метотрексата осуществляются один раз в неделю, с одновременным введением цитозара, преднизолона. В конце полихимиотерапии подключается облучение головы.
Поддерживающая терапия проводится двумя препаратами сроком до 2-х лет .
6-Меркаптопурин 50 мг/м 2 внутрь, ежедневно.
Метотрексат 20 мг/м 2 внутрь 1 раз в неделю. В день приема необходимо контролировать общий анализ крови и регулировать дозу поддерживающей терапии в зависимости от уровня лейкоцитов (при уровне лейкоцитов 1-2 * 10 9 /л – 50% доза 6 МП/МТХ, 2-3 * 10 9 /л – 100%, более 3 * 10 9 /л – 150 %).
В республиканской детской больнице выживаемость детей с ОЛЛ была около 10-12%. 1990 г., после начала работы по протоколам БФМ выживаемость детей с первичным ОЛЛ составила 90%, ремиссия 95%, 5 -летняя выживаемость 69%.
После изложения основной терапии по протоколам БМФ – 90 необходимо остановиться на клиническом патоморфозе лейкозов.
Само понятие “патоморфоз” (греч.) – стойкое изменение количественных и качественных сдвигов нозологии под влиянием в данном случае лечения: стойкое изменение лимфобластных лейкозов.
К проявлениям патоморфоза относится поражение органов лейкозным процессом с присущими ему интоксикацией, инфильтрацией, кровоизлияниями и анемической гипоксией, а также побочным действием цитостатиков. В процессе лечения с наступлением ремисси редуцируется лейкозная опухоль и уходят ее проявления в органах и системах, но сохраняются побочные действия цитостатиков, ибо после наступления ремиссии продолжается специфическая химио- и лучевая терапия .
Самым важным звеном в этом патоморфозе является цитопенический синдром, когда разрушены бласты и нормальное кроветворение еще не восстановилось. В этом опустошении без кроветворных клеток организм подстеригает череда осложнений: синдром разрушения бластов (знаменитая тетрада – гиперурекимия, гиперкалиемия, гиперфосфатемия, гипокальциемия), ДВС-синдром, сепсис и кровоизлияния, поражение сердца, печени, кишечника и др. органов. В этот период даже при отсрочивании назначений цитостатиков, действие их на организм еще продолжается. Все это создает сложную ситуацию, в которой необходимо предусмотреть помощь всему организму в целом, поэтому большое значение при лечении ОЛЛ имеет, так называемая, сопроводительная терапия.
С целью противодействия синдрому лизиса опухоли, с первых дней лечения проводится массивная инфузионная терапия (3000 мл/кв.м в сутки) глюкозо-солевыми растворами. Для профилактики и лечения инфекционных осложнений широко используются противовирусные препараты Зовиракс (ацикловир), Вальтрекс Glaxo Wellcome и др., антибиотики цефалоспорины П-го поколения (Зинацеф, Glaxo Wellcome) и Ш-го поколения (Фортум, Glaxo Wellcome,Роцефин, F.Hoffmann-La Roche и др.), аминогликозиды и др., антиникотические средства — дифлюкан, амфотерицин.
Адекватным антиперетиком является парацетамол (Gllaxo Wellcome). Одним из наиболее эффективных антиэметических препаратов является Зофран (Glaxo Wellcome). Арсенал препаратов, используемых в качестве сопроводительной терапии при ОЛЛ у детей очень велик.
Хронический миелолейкоз (ХМЛ) – это опухоль кроветворной ткани, возникающая из клеток-предшественниц миелопоэза, сохраняющих способность дифференцироваться до зрелых форм. Хронический лейкоз отличается от острого не длительностью течения, а морфологическим субстратом опухоли. Основной морфологический субстрат ХМЛ составляют дифференцирующиеся и зрелые гранулоциты.
Опухолевый процесс при ХМЛ проходит две стадии:
1. Развернутую – моноклоновую, доброкачественную (зрелоклеточную), характеризующуюся длительностью течения.
2. Терминальную – поликлоновую, злокачественную (в терминальной стадии субстрат опухоли составляют бласты), значительно более короткую.
ХМЛ является опухолью, имеющей четкие хромосомные нарушения – вместо нормальной хромосомы из 22-й пары хромосому с укороченным длинным плечом – Ph 1 -хромосому (“филадельфийскую”), наличие которой считается патогмоничным признаком ХМЛ. В развернутой стадии заболевания 90% и более клеток содержат Ph 1 -хромосому. Она определяется в делящихся клетках миелоидного, эритроидного и мегакариоцитарного ряда, моноцитарно-макрофагальных элементах, В-лимфоцитах. Наличие маркерной хромосомы в этих клетках позволяет считать, что все они происходят из полипотентной стволовой клетки.
ХМЛ встречается в любом возрасте, но в отличие от острого лейкоза, редко (2-5,5% от всех лейкозов).
У детей выделяют две формы ХМЛ: взрослую и ювенильную (инфантильную), различающихся по клинико-гематологическим проявлениям, течению заболевания, наличию Ph 1 -хромосомы, ответной реакции на терапию.
Взрослая форма ХМЛ (ВХМЛ) встречается у детей чаще, чем ювенильная. В основном ВХМЛ наблюдается у детей школьного возраста. Ее клинико-гематологические проявления напоминают таковые у взрослых, чем и объясняется название этой формы. ВХМЛ начинается постепенно. Больные длительно не предъявляют жалоб и в начальную стадию болезнь обнаруживается случайно при обследовании по поводу других заболеваний или при оформлении в детские учреждения, когда выявляются изменения в периферической крови (лейкоцитоз со сдвигом влево до мета- и миелоцитов), иногда уже увеличивается селезенка. Миелограмма – без патологических отклонений. Патогмонично обнаружение клеток, содержащих Ph 1 -хромосому.
Как правило, болезнь диагностируется в развернутой стадии. В связи с постепенно нарастающей интоксикацией появляются жалобы на общую слабость, недомогание, ухудшение аппетита, похудание, повышение температуры, могут быть боли в животе, оссалгии, незначительная аденопатия, единичные геморрагии на теле. Кожа приобретает бледно-серый оттенок. Характерна спленомегалия, за счет которой изменяется конфигурация живота, появляется чувство распирания, иногда боли (за счет перерастяжения капсулы селезенки, переспленита), возможно развитие инфаркта селезенки. Селезенка плотная, может увеличиваться до таких размеров, что занимает пространство до области таза. Печень увеличена меньше.
Для периферической крови в развернутой стадии ВХМЛ характерен нейтрофильный гиперлейкоцитоз, сдвиг влево до миело- и промиелоцитов, встречаются единичные миелобласты, увеличивается количество базофилов и эозинофилов (“базофильно-эозинофильная ассоциация”). Почти у всех больных отмечается умеренная нормохромная анемия. Несмотря на нормальное и даже повышенное количество тромбоцитов наблюдается нарушение их функций, т.е. имеется дефицит “пула хранения”. СОЭ обычно увеличена. Костный мозг гиперпластичен, миелоидные клетки находятся на всех стадиях дифференцировки, л:э может составлять 10:1 – 20:1. Диагностически ценны результаты исследования костного мозга с изучением хромосом (обнаружение Ph 1 -хромосомы). Характерно также снижение активности щелочной фосфатазы (ЩФ) в нейтрофилах вплоть до полного ее отсутствия.
Клинические и гематологические проявления терминальной стадии (бластного криза ВХМЛ) отличаются от развернутой стадии и практически не отличаются от симптоматики острого лейкоза (ОЛ). Как правило, бластный криз развивается на фоне лечения ВХМЛ. К ранним предвестникам его относятся нарастающий лейкоцитоз и увеличение селезенки, что свидетельствует о развитии рефрактерности к проводимой терапии. У больного ухудшается самочувствие, нарастают слабость, потливость, бледность, появляется и прогрессирует геморрагический синдром, нередко повышение температуры до 38-40 0 С, некротические поражения кожи и слизистых, выраженный болевой синдром (боли в животе, спине, ногах, припухлость и боли в суставах). Увеличиваются селезенка, печень. У некоторых больных терминальная стадия может проявиться в виде экстрамедуллярных опухолевых образований (увеличения лимфоузлов, остеолитических изменений, нейролейкоза, лейкемидов в коже). При исследовании этих образований выявляются бласты, цитогенетически определяется Ph-хромосома. Экстрамедуллярная бластная трансформация может предшествовать бластной трансформации костного мозга за несколько месяцев. У некоторых больных заболевание может сразу проявиться бластным кризом. Следует отметить, что бластный криз может протекать по миелоидному, лимфоидному, смешанному типу, т.е. в период бластного криза ВХМЛ возможна пролиферация различных клеточных линий. Это также свидетельствует о том, что поражение происходит на уровне полипотентной стволовой клетки.
В периферической крови – нарастающая нормохромная анемия, выраженная тромбоцитопения, гиперлейкоцитоз с появлением в лейкоцитарной формуле большого количества бластов (суммарное содержание бластов и промиелоцитов > 30% свидетельствует в пользу бластного криза). Картина периферической крови становится похожей на таковую при остром миелобластном лейкозе. В миелограмме характерно нарастание бластных клеток; однако, бласты в периферической крови преобладают над бластами в костном мозге.
Ювенильная форма ХМЛ (ЮХМЛ) по частоте возникновения занимает 2-е место после “взрослого” типа ХМЛ и наблюдается преимущественно у детей до 2-х лет. Клинические проявления ЮХМЛ напоминают острый лейкоз. При ЮХМЛ трудно выделить и разграничить отдельные периоды заболевания в связи с быстрой распространенностью процесса. Появляется лихорадка, быстро нарастают симптомы интоксикации, анемический и геморрагический синдромы, увеличиваются печень и селезенка, нередки оссалгии, некротические поражения кожи, язвенно-некротический стоматит, значительно увеличиваются все группы лимфоузлов.
В периферической крови выражены нормохромная анемия, тромбоцитопения, лейкоцитоз. Лейкоцитарная формула напоминает таковую при ВХМЛ: имеются все переходные формы нейтрофильного ряда, но преобладают зрелые формы. Характерен моноцитоз, отсутствует “базофильно-эозинофильная ассоциация”. СОЭ увеличена. В эритроцитах резко повышено содержание НвF (30-70% и более), отмечается гипергаммаглобулинемия с превалированием IgG. В миелограмме в основном определяются клетки миелоидного ряда с увеличением элементов митотического пула, количество бластов нормальное или незначительно повышено (до 10%), соответствуя таковому числу бластов в периферической крови.
Ph 1 -хромосома отсутствует. По данным разных авторов у 50-82% детей с ЮХМЛ отмечается нормальный кариотип, однако нередко у больных в костномозговых клетках обнаруживались различные цитогенетические аномалии.
Диагностические критерии взрослой и ювенильной форм ХМЛ.
источник