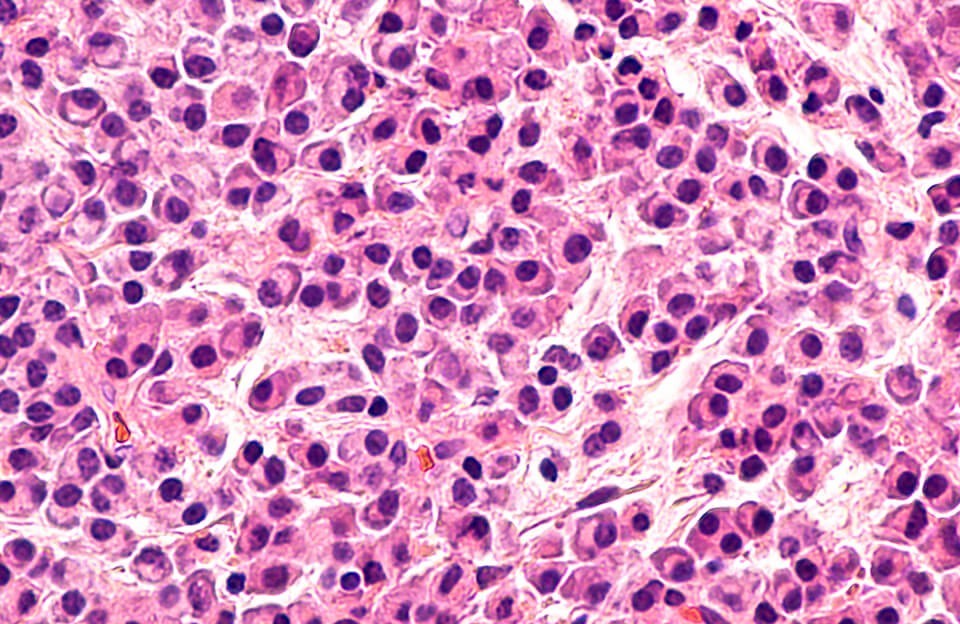
Это опухолевое заболевание, возникающее из плазматических клеток крови (подтип лейкоцитов, т.е. белых кровяных телец). У здорового человека эти клетки участвуют в процессах иммунной защиты, вырабатывая антитела. При миеломной болезни (ее еще называют миеломой) в костном мозге и в костях накапливаются измененные плазматические клетки, что нарушает образование нормальных клеток крови и структуру костной ткани. Иногда можно услышать о миеломе костей, миеломной болезни позвоночника, почек или крови, но это не совсем верные названия. Миеломная болезнь уже подразумевает поражение кроветворной системы и костей.
Болезнь неоднородна, можно выделить следующие варианты:
моноклональная гаммапатия неясного генеза — это группа заболеваний, при которых избыточное количество В-лимфоцитов (это клетки крови, которые участвуют в иммунных реакциях) одного вида (клона) вырабатывают аномальные иммуноглобулины разных классов, которые накапливаются в разных органах и нарушают их работу (очень часто страдают почки).
лимфоплазмацитарная лимфома (или неходжкинская лимфома), при которой очень большое количество синтезирующихся иммуноглобулинов класса М повреждает печень, селезенку, лимфоузлы.
плазмацитома бывает двух видов: изолированная (поражает только костный мозг и кости) и экстрамедуллярная (накопление плазматических клеток происходит в мягких тканях, например, в миндалинах или носовых пазухах). Изолированная плазмацитома костей в некоторых случаях переходит во множественную миелому, но не всегда.
Множественная миелома составляет до 90% всех случаев заболевания и обычно затрагивает несколько органов.
асимптоматическую (тлеющую, бессимптомную миелому)
миелому с анемией, поражением почек или костей, т.е. с симптомами.
Стадии определяют в зависимости от количества в сыворотке крови бета-2 микроглобулина и альбумина.
1 стадия миеломы: уровень бета-2 микроглобулина менее 3,5 мг/л, а уровень альбумина равен 3,5 г/дл или более.
2 стадия миеломы: бета-2 микроглобулин колеблется между 3,5 мг/л и 5, 5 мг/л или же уровень альбумина ниже 3,5, тогда как бета-2 микроглобулин ниже 3,5.
3 стадия миеломы: уровень бета-2 микроглобулина в сыворотке составляет более 5,5 мг/л.
Причины миеломы неизвестна. Есть ряд факторов, повышающих риск заболеть:
Возраст. До 40 лет миеломной болезнью почти не болеют, после 70 лет риск развития заболевания значительно повышен
Мужчины болеют чаще женщин
У людей с черным цветом кожи риск миеломы в два раза выше, чем у европейцев или азиатов
Имеющаяся моноклональная гаммапатия. У 1 из 100 человек гаммапатия трансформируется в множественную миелому
Семейная история заболевания миеломой или гаммапатией
Патология иммунитета (ВИЧ или применение лекарств, подавляющих иммунитет)
Воздействие радиации, пестицидов, удобрений
Костный мозг в обычных условиях вырабатывает строго определенное количество В-лимфоцитов и плазматических клеток. При миеломной болезни их выработка выходит из-под контроля, костный мозг заполняется аномальными плазматическими клетками, а образование нормальных лейкоцитов и эритроцитов снижается. При этом вместо антител, полезных в борьбе с инфекциями, такие клетки производят белки, способные повреждать почки.
Признаки, которые помогут заподозрить миелому:
Боль в костях, особенно в ребрах и позвоночнике
Патологические переломы костей
Частые, повторяющиеся случаи инфекционных заболеваний
Выраженная общая слабость, постоянная усталость
Кровотечения из десен или носовые, у женщин — обильные менструации
Головная боль, головокружение
Поставить диагноз бывает сложно, так как при миеломной болезни нет какой-то явной опухоли, которую можно заметить, а иногда заболевание вообще протекает без каких-либо симптомов.
Диагностикой миеломной болезни обычно занимается врач-гематолог. Во время расспроса врач выявляет основные симптомы заболевания у данного пациента, выясняет, нет ли кровотечений, боли в костях, частых простуд. Затем проводят дополнительные исследования, необходимые для точной постановки диагноза и определения стадии болезни.
Клинический анализ крови при миеломе часто указывает на увеличение вязкости крови и повышение скорости оседания эритроцитов (СОЭ). Нередко снижено количество тромбоцитов и эритроцитов, гемоглобина.
В результатах анализа крови на электролиты часто повышен уровень кальция; по данным биохимического анализа увеличено количество общего белка, определяются маркеры нарушения функции почек — высокие цифры мочевины, креатинина.
Анализ крови на парапротеин проводят для оценки типа и количества аномальных антител (парапротеинов).
В моче часто определяют патологический белок (белок Бенс-Джонса), который представляет собой моноклональные легкие цепи иммуноглобулинов.
На рентгенограмме костей (черепа, позвоночника, бедренных и тазовых костей) видны характерные для миеломы повреждения.
Пункция костного мозга — самый точный способ диагностики. Тонкой иглой берут кусочек костного мозга, обычно прокол делают в области грудины или кости таза. Затем полученный изучают под микроскопом в лаборатории на предмет перерожденных плазматических клеток и проводят цитогенетическое исследование для выявления изменений в хромосомах.
Компьютерная томография, магнитно-резонансная томография, ПЭТ-сканирование позволяют выявить участки повреждения в них.
В настоящее время используют разные методы лечения, прежде всего лекарственную терапию, при которой препараты применяют в разных комбинациях.
Таргетная терапия с использованием лекарства (бортезомиб, карфилзомиб (не зарегистрирован в России), которые за счет влияния на синтез белков вызывают гибель плазматических клеток.
Терапия биологическими препаратами, такие как талидомид, леналидомид, помалидомид, стимулируют собственную иммунную систему бороться с опухолевыми клетками.
Химиотерапия циклофосфаном и мелфаланом, которые тормозят рост и приводят к гибели быстрорастущих клеток опухоли.
Кортикостероидная терапия (дополнительное лечение, которое усиливает эффект основных препаратов).
Бисфосфонаты (памидронат, золендроновая кислота) назначают для повышения плотности костной ткани.
Обезболивающие, в том числе наркотические анальгетики, применяют при сильных болях (очень частая жалоба при миеломной болезни), для облегчения состояния пациента применяют хирургические методы и лучевая терапия.
Хирургическое лечение требуется, например, для фиксации позвонков с помощью пластин или других приспособлений, так как происходит разрушение костной ткани, в том числе позвоночника.
После проведения химиотерапии часто проводят пересадку костного мозга, при этом наиболее эффективной и безопасной является аутологичная трансплантация стволовых клеток костного мозга, Для осуществления этой процедуры проводят забор стволовых клеток красного костного мозга. затем назначают химиотерапию (как правило, высокими дозами противоопухолевых препаратов), которая уничтожает раковые клетки. После окончания полного курса лечения делают операцию по пересадке забранных ранее образцов, и в результате начинают расти нормальные клетки красного костного мозга.
Некоторые формы заболевания (прежде всего “тлеющая” меланома) не требуют срочного и активного лечения. Химиотерапия вызывает тяжелые побочные эффекты и в некоторых случаях — осложнения, а эффекты на течение болезни и прогноз при бессимптомной “тлеющей” миеломной болезни сомнителен. В таких случаях проводят регулярное обследование и при первых признаках обострения процесса начинают лечение. План контрольных исследований и регулярность их проведения врач устанавливает индивидуально для каждого пациента, и очень важно соблюдать эти сроки и все рекомендации врача.
Сильные боли в костях, требующие назначения эффективных обезболивающих средств
Почечная недостаточность с необходимостью гемодиализа
Частые инфекционные заболевания, в т.ч. воспаление легких (пневмонии)
Истончение костей с переломами (патологические переломы)
Анемия, требующая переливания крови
При “тлеющей” миеломе заболевание может не прогрессировать десятки лет, но необходимо регулярное наблюдение у врача, чтобы вовремя заметить признаки активизации процесса, при этом появление очагов разрушения костей или увеличение в костном мозге количества плазматических клеток выше 60% говорит об обострении заболевания (и ухудшении прогноза).
Выживаемость при миеломе зависит от возраста и общего состояния здоровья. В настоящее время в целом прогноз стал оптимистичнее, чем еще 10 лет назад: 77 из 100 больных миеломой человек будут жить как минимум год, 47 из 100 — как минимум 5 лет, 33 из 100 — как минимум 10 лет.
Причины смерти при миеломной болезни
Чаще всего к смерти приводят инфекционные осложнения (например, пневмония), а также фатальные кровотечения (связанные с низким количеством тромбоцитов в крови и нарушениями свертываемости), переломы костей, тяжелая почечная недостаточность, тромбоэмболия легочной артерии.
Питание при миеломной болезни
Рацион при миеломе должен быть разнообразным, содержать достаточное количество овощей и фруктов. Рекомендуют уменьшить потребление сладостей, консервов и готовых полуфабрикатов. Специальной диеты можно не придерживаться, но так как миеломе часто сопутствует анемия, то желательно регулярно есть продукты, богатые железом (постное красное мясо, сладкий перец, изюм, брюссельская капуста, брокколи, манго, папайя, гуава).
В одном из исследований было продемонстрировано, что употребление куркумы предупреждает резистентность к химиотерапии. Исследования на мышах показали, что куркумин может замедлять рост раковых клеток. Также добавление в пищу куркумы во время химиотерапии может несколько облегчить тошноту и рвоту.
Все изменения в диете нужно согласовывать с лечащим врачом, особенно во время химиотерапии.
источник
Множественная миелома (ММ), ранее обозначаемая как миеломная болезнь или плазмоцитома, – опухоль, возникающая на уровне ранних предшественников β-лимфоцитов, при этом моноклональный пул потомков первично трансформированной клетки сохраняет способность к дифференцировке до конечного этапа – плазматических клеток, секретирующих иммуноглобулины. Следовательно, субстратом опухоли служат плазматические клетки. С этим связано и более раннее ее название – плазмоцитома. Так как опухоль продуцирует патологический иммуноглобулин – парапротеин, то ее относят к группе парапротеинемических гемобластозов. Учитывая то, что она происходит из ранних предшественников β-лимфоцитов, ее относят к группе лимфопролиферативных заболеваний.
Заболеваемость в среднем составляет 50 случаев в год на 1 млн населения. ММ представлено 15% всех случаев лимфоидных злокачественных новообразований и 2% всех типов злокачественных опухолей. Мужчины болеют немного чаще женщин. Пик заболеваемости приходится на возраст около 67–71 года. В молодом возрасте (до 40 лет) ММ регистрируют крайне редко. Случаи заболевания детей не зарегистрированы.
Причины заболевания, как и этиология опухолей вообще, неизвестны.
В основе заболевания лежит пролиферация плазматических клеток. Плазмоцит (плазматическая клетка) происходит из коротко живущих β-лимфоцитов и обладает способностью вырабатывать неограниченное количество антител, специфических практически для любого антигена. При ММ все клетки, составляющие массу опухоли, происходят из одной клетки клона, потомки которой повторяют функцию клетки-родоначальницы и в большом количестве секретируют иммуноглобулин лишь одной структуры (моноклоновый иммуноглобулин). Количество нормальных плазматических клеток уменьшается, как и содержание нормальных иммуноглобулинов, выполняющих функцию антител. В связи с этим возникает иммунодефицитное состояние, способствующее развитию инфекционных осложнений. Продуцируемый лейкозными β-лимфоцитами и (или) связанными с ними дополнительными клетками ИЛ-6 потенцирует рост и развитие плазматических клеток, а другие высвобождаемые цитокины (ФНО-а и ИЛ-1) ускоряют резорбцию костной ткани. При генетическом анализе обнаруживают мутации в онкогенах и транслокации в хромосомах.
Первоначально опухоль локализуется в костном мозге. В дальнейшем опухолевые клетки (плазмоциты) метастазируют в органы (селезенку, печень). Увеличенное количество плазматических клеток в костном мозге в дальнейшем вытесняет эритробластический и миелоцитарный ростки костного мозга.
В основу современной классификации положены объем опухолевой ткани (стадии течения) и активность патологического процесса (степень агрессивности гемобластоза).
1. I стадия (малая масса опухоли): концентрация гемоглобина более 100 г/л, нормальное содержание кальция в крови, нет остеолиза или очагового поражения костей, низкая концентрация IgM. Содержание IgG менее 50 г/л, IgA – менее 30 г/л. Выведение белка Бен-Джонса – менее 4 г/сут. Содержание креатинина в крови не увеличено.
2. II стадия (средняя масса опухоли): показатели средние между таковыми в I и III стадии болезни.
3. III стадия (большая масса опухоли): концентрация гемоглобина менее 85 г/л, содержание кальция в крови выше нормы, выраженный остеодеструктивный процесс, высокая концентрация IgM при содержании IgG более 70 г/л, IgA – более 50 г/л. Экскреция белка Бен-Джонса с мочой – 12 г/сут. Содержание креатинина в крови повышено.
Активность патологического процесса определяют следующим образом:
1. «тлеющая» ММ (малоагрессивная) – признаки прогрессирования отсутствуют в течение многих месяцев или лет;
2. медленно прогрессирующая;
3. быстро прогрессирующая (агрессивная).
Все эти показатели помогают не только оценить особенности патологического процесса, но и позволяют подобрать оптимальное лечение.
Анатомически (на основании данных рентгенологического исследования скелета и цитологического и патоморфологического анализа пунктатов и трепанатов костей) выделяют следующие формы ММ:
1. диффузно-очаговую (наиболее распространенную, около 60% больных);
3. множественно-очаговую (15%);
4. редкие формы (склерозирующая, преимущественно висцеральная – 1%).
Выделение анатомических форм оправдано с точки зрения возможности получения при первой же стернальной пункции субстрата болезни (увеличенного числа плазматических клеток).
Симптомы заболевания определяют несколько больших синдромов – костномозговой, белковых нарушений и висцеральный.
Костномозговой синдром обусловлен пролиферацией в костном мозге миеломных клеток, что приводит к разрушению костного вещества. В первую очередь деструктивные процессы (остеопороз, остеолиз) развиваются в плоских костях и позвоночнике. Иногда первые очаги разрушения обнаруживают в проксимальных отделах трубчатых костей. Гиперплазия костного мозга вследствие разрастания скоплений миеломных клеток также приводит к вытеснению миелоидных элементов.
В результате перечисленных процессов развиваются:
1. остеопороз, патологические переломы, гиперкальциемия;
2. анемия, лейкопения, тромбоцитопения (реже) в периферической крови;
3. миеломноклеточная метаплазия в костном мозге.
Синдром белковых нарушений обусловлен гиперпродукцией моноклонового парапротеина плазматическими клетками, уменьшением секреции нормальных иммуноглобулинов и представлен следующими признаками:
3. геморрагическим диатезом;
4. синдромом повышенной вязкости;
5. периферической невропатией;
6. синдромом недостаточности антител (с развитием инфекционных осложнений).
Миелоидная нефропатия – наиболее частый и серьезный симптом парапротеинемии. Она приводит к почечной недостаточности, которая занимает одно из первых мест среди причин смерти больных. В основе развивающейся почечной недостаточности лежит нефросклероз. Его причиной служит реабсорбция в канальцах белка, в большом количестве фильтрующегося в клубочках. Это связано с тем, что за счет парапротеина в крови значительно увеличено содержание белка. Реабсорбируемый парапротеин инфильтрирует ткань почки, способствуя развитию склероза. Доказано раннее вовлечение в патологический процесс базальной мембраны капилляров нефрона и мезангиума с их последующим склерозированием. Клинические признаки миелоидной нефропатии складываются из упорной (иногда многолетней) протеинурии и постепенно развивающейся ХПН. Особенность поражения почек – отсутствие отеков и симптомов сосудистого поражения (АГ, ретинопатии).
Амилоидоз LA-типа – тканевый парапротеиноз, регистрируемый в 15% случаев. В отличие от классического вторичного амилоидоза отмечают поражение органов, богатых коллагеном: сосудов (адвентиции), сердца, языка, суставов и сухожилий. Печень, селезенка и почки не страдают. Параамилоидоз не всегда сопровождается клиническими симптомами и часто бывает лишь патологоанатомической находкой. Тем не менее в ряде случаев можно обнаружить макроглоссию, прогрессирующую сердечную недостаточность, а также упорные боли в суставах с их деформацией. Прижизненная диагностика параамилоидоза затруднена; необходима биопсия кожи, слизистых оболочек (рта, прямой кишки), лимфатических узлов и мышц.
Геморрагический синдром – редкое явление. Кровоточивость из сосудов слизистых оболочек и кожи обусловлена тем, что парапротеин, «окутывая» тромбоциты, затрудняет их адгезию и агрегацию.
Синдром повышенной вязкости – нарушение микроциркуляции вследствие высокой гиперпротеинемии – манифестирует геморрагической ретинопатией, расширением вен сетчатки и нарушениями периферического кровотока вплоть до акрогангрены. При охлаждении тела эти явления могут усиливаться вследствие выпадения криоглобулинов.
Периферическую невропатию регистрируют в 5% случаев. Она выражается в нарушениях тактильной и болевой чувствительности, а также в парестезиях. При гистологическом исследовании обнаруживают дегенеративные изменения нервных волокон.
Синдром недостаточности антител обусловлен резким снижением концентрации нормальных иммуноглобулинов вплоть до их полного исчезновения. Вторичная гипогаммаглобулинемия приводит к выраженной склонности к инфекционным осложнениям, особенно со стороны мочевыводящих путей и бронхолегочного аппарата.
Висцеральный синдром заключается в лейкемической инфильтрации внутренних органов (главным образом, печени и селезенки). В 5–12% случаев при жизни больных обнаруживают гепато- и спленомегалию. Опухолевые плазмоклеточные инфильтраты можно обнаружить практически во всех внутренних органах, но они редко манифестируют клинически и обычно служат патологоанатомическими находками.
Различная выраженность перечисленных синдромов и степени нарушений белкового обмена обусловливает чрезвычайную вариабельность течения болезни. Можно наблюдать пациентов с несомненной ММ, предъявляющих небольшое число жалоб или вообще не отмечающих никаких нарушений. В то же время есть больные, нуждающиеся в проведении постоянного лечения и утратившие трудоспособность вследствие тяжелой инвалидности, связанной с патологическими переломами (прежде всего компрессионными переломами позвоночника).
Заболевание можно обнаружить на разных стадиях течения, но у ряда больных (особенно среди тех, у кого ММ диагностировали рано) можно выделить две стадии болезни:
1. относительно доброкачественную, характеризующуюся соматической компенсацией, отсутствием или медленным прогрессированием остеодеструктивного процесса, нормальными показателями крови, стабильно невысоким содержанием патологического иммуноглобулина (парапротеина) и сохранностью нормальных иммуноглобулинов;
2. быстро прогрессирующую, при которой нарастает разрушение костей, возникают метастазы во внутренние органы, концентрация парапротеина резко повышается, а содержание нормальных иммуноглобулинов резко снижается вплоть до развития выраженной гипогаммаглобулинемии, при этом развиваются анемия, лейкопения и повышается количество плазмобластов.
Все вышесказанное обусловливает получение самых разных данных на всех этапах диагностического поиска.
На первом этапе диагностического поиска в начальной стадии болезни больные могут не предъявлять жалоб, и болезнь диагностируют после соответствующего обследования на основании случайного обнаружения протеинурии или значительного увеличения СОЭ (диспансеризация, обращение к врачу по иным причинам), что обычно отмечают в 20% случаев.
Больные могут замечать, что у них в течение многих лет обнаруживают увеличение СОЭ (иногда довольно значительное – до 50–60 мм/ч), при этом тщательное обследование, как правило, направленное на определение злокачественной опухоли, не обнаруживало причины болезни. Как правило, стернальную пункцию или трепанобиопсию не проводили. В половине случаев болезнь дебютирует слабостью, повышенной утомляемостью, снижением массы тела и болями в костях. Иногда заболевание сразу же манифестирует сильными болями в костях или переломами ребер, гребней подвздошных костей, а также компрессионными переломами позвонков. Часто больные страдают вялотекущими пневмониями, которые нередко рецидивируют и плохо поддаются лечению антибиотиками. Также регистрируют заболевания мочевыводящих путей (циститы, пиелиты), характеризующиеся дизурическими расстройствами и упорным субфебрилитетом.
В анамнезе у больных могут быть указания на ранее проводимое лечение цитостатическими препаратами, а также сеансы плазмафереза, после чего их состояние улучшалось.
На втором этапе диагностического поиска в начальных стадиях болезни нередко не обнаруживают никаких патологических изменений. В развернутой стадии заболевания иногда обнаруживают нарушения, обусловленные вышеуказанными синдромами (костномозговым, висцеральным, белковых нарушений). При вялотекущих пневмониях можно отметить участки укорочения перкуторного звука и стойких влажных звонких мелкопузырчатых хрипов. Как правило, обнаруживают болезненность при поколачивании плоских костей. При их деструкции (патологические переломы) образуются участки резкой болезненности и нарушается функция пораженной кости. Неспецифические симптомы – снижение массы тела, субфебрилитет и повышенная потливость.
Следует отметить, что необнаружение симптомов, обусловленных вышеперечисленными синдромами, на втором этапе диагностического поиска отнюдь не отвергает предположение о ММ, но свидетельствует об отсутствии грубых изменений со стороны пораженных органов и систем.
Третий этап диагностического поиска считают решающим для установления диагноза.
При исследовании периферической крови не обнаруживают специфических признаков. У всех больных по мере прогрессирования заболевания развивается анемия, патогенез которой, вероятно, связан с вытеснением нормального кроветворения растущей опухолью. Тем не менее прямой зависимости между степенью анемии и выраженностью костных поражений нет.
Число лейкоцитов и лейкоцитарная формула обычно не изменены. Иногда отмечают нейтропению с относительным лимфоцитозом, реже – умеренный нейтрофилез со сдвигом лейкоцитарной формулы влево и появлением молодых форм гранулоцитарного ряда. При прогрессировании заболевания обнаруживают выраженную лейко- и нейтропению (особенно при лечении цитостатическими препаратами). Часто регистрируют абсолютный моноцитоз.
Мегакариоцито- и тромбоцитопоэз долгое время не изменены. На ранних стадиях иногда отмечают гипертромбоцитоз и увеличение числа мегакариоцитов в пунктате костного мозга.
Характерно значительное увеличение СОЭ (до 60–80 мм/ч).
При анализе миелограммы обнаруживают отчетливую миеломноклеточную пролиферацию: количество миеломных опухолевых клеток превышает 10%, часто отмечают патологические клетки – многоядерные или аномальной формы.
Если диффузного поражения костного мозга нет (лишь «гнездное» поражение), то миелограмма может соответствовать норме. В этой ситуации при подозрении на плазмоцитому (остеолитические очаги, моноклональная иммуноглобулинопатия) необходимо проводить повторные проколы грудины в разных участках, пунктировать или трепанировать гребни подвздошной кости, проводить пункции в местах остеолитических дефектов или костных опухолей.
При биохимическом исследовании закономерно регистрируют гиперпротеинемию: содержание общего белка достигает 10–12 г/л. При электрофоретическом исследовании обнаруживают дополнительную фракцию (М-градиент) в области γ-глобулиновой фракции, при этом количество нормальных γ-глобулинов резко снижено. Эта дополнительная фракция служит отражением высокой концентрации парапротеина в крови. При исследовании содержания иммуноглобулинов отмечают резкое увеличение концентрации какого-либо класса – IgA, G, Е или D, но не IgM, что свойственно макроглобулинемии Вальденстрема – другому парапротеинемическому гемобластозу, обусловленному гиперплазией короткоживущих β-лимфоцитов. Часто в крови повышено содержание в2-макроглобулина. Чем выше концентрация этого белка, тем хуже прогноз у пациента.
При иммуноэлектрофорезе удается провести более детальное типирование парапротеина при миеломной болезни: определяют класс тяжелых цепей парапротеина – A, G, Е или D, а также тип легких цепей – к (каппа) или ? (лямбда). Возможно развитие особого варианта ММ – так называемой миеломы БенДжонса, парапротеин которой состоит лишь из легких цепей (микромолекулярный вариант болезни).
В моче достаточно часто можно определить разной степени протеинурию. При миеломе Бен-Джонса в ней присутствует одноименный белок. Нагревание мочи приводит к его выпадению в осадок, а дальнейшее нагревание – к растворению.
При рентгенологическом исследовании костей можно обнаружить изменения плоских костей (особенно костей черепа) в виде круглых просветлений в костной ткани, представляющих участки ее резорбции. Можно также отметить переломы костей, особенно компрессионные переломы тел позвонков.
Следует помнить, что не существует специфических изменений скелета, характерных для ММ. Отсутствие остеодеструкции не исключает ММ, а ее обнаружения недостаточно для установления диагноза, так как для этого необходимы другие признаки, о которых будет сказано ниже.
Гиперкальциемию регистрируют в 20–40% случаев, чаще – в терминальной стадии болезни (особенно при ХПН).
При ХПН присутствуют все ее лабораторные признаки: снижение плотности мочи, уменьшение СКФ и увеличение концентрации креатинина в крови.
Для установления диагноза ММ используют две группы критериев.
• плазмоклеточная инфильтрация костного мозга (по данным трепанобиопсии);
• увеличение количества плазматических клеток в миелограмме более 35%;
• концентрация IgG более 35 г/л, IgA – свыше 20 г/л (по данным электрофореза сыворотки крови), содержание легких цепей иммуноглобулинов при отсутствии признаков амилоидоза более 1,0 г в суточном объеме мочи (по данным электрофореза мочи).
• содержание плазматических клеток в костном мозге около 10–30%;
• присутствие моноклонального иммуноглобулина (по данным электрофореза сыворотки крови), но в меньшей концентрации;
• обнаружение очагов остеолиза;
• содержание иммуноглобулинов, не превышающее для IgM 0,5 г/л, для IgA – 1 г/л, для IgG – 6 г/л.
Диагноз ММ устанавливают при обнаружении одного большого и одного малого или одного большого и двух малых критериев (1+1 или 1+2).
Трудности в диагностике ММ возникают на ее ранних стадиях, когда отсутствует костная деструкция, нет отчетливой миеломноклеточной метаплазии костного мозга, невелик М-градиент при электрофорезе белков сыворотки и нет выраженного снижения содержания γ-глобулинов. Эти стадии течения ММ неотличимы от так называемых эссенциальных (при беременности, у лиц пожилого возраста) и симптоматических (при циррозе печени, диффузных заболеваниях соединительной ткани, злокачественных опухолях, сепсисе) моноклональных гаммапатий. Тщательное исследование позволяет исключить реактивную гаммапатию. Этому также способствует динамическое наблюдение за больными. Следует помнить, что на ранних стадиях болезни, когда больной попадает в поле зрения врача, правильный диагноз может быть поставлен через несколько лет после обнаружения парапротеина в крови.
ММ необходимо дифференцировать от ряда заболеваний и состояний.
1. Макроглобулинемия Вальденстрема – одна из опухолей лимфатической системы, рассматриваемая в рамках парапротеинемических гемобластозов, так как речь идет о пролиферации в системе лимфоцитов (источник продукции IgM). Этим заболеванием страдают преимущественно мужчины (до 70%) в возрасте около 60 лет. Клиническая картина чрезвычайно сходна с ММ и обусловлена лейкемической пролиферацией лимфоидных элементов костного мозга, печени, селезенки и лимфатических узлов, накоплением в сыворотке крови парапротеина, тяжелую цепь которого относят к М-классу. Костно-деструктивный процесс развивается редко, болевой синдром обычно отсутствует. Характерна гепато- и спленомегалия. Увеличение печени, селезенки и лимфатических узлов связано с разрастанием лимфатических элементов. Картина костного мозга характеризуется увеличением лимфоцитов, но повышено и количество плазматических клеток. Все остальные синдромы при макроглобулинемии Вальденстрема достаточно выражены, но в отличие от ММ поражение почек обнаруживают редко, что, вероятно, связано с отсутствием гиперпротеинемии и протеинурии. Главное отличие макроглобулинемии Вальденстрема от ММ состоит в обнаружении парапротеина класса IgM.
2. Доброкачественная моноклоновая гаммапатия (синоним: моноклоновая гаммапатия неясной этиологии) – вялотекущее заболевание, характеризующееся стабильным и относительно невысоким (менее 20 г/л) содержанием парапротеина в сыворотке крови. Это состояние регистрируют чаще ММ. Концентрация нормальных иммуноглобулинов не снижена. Отсутствуют поражение костей и белок Бен-Джонса в моче, а число плазматических клеток в костном мозге обычно не превышает 10%. У 10–30% больных это заболевание медленно трансформируется в ММ или лимфому.
3. При первичном амилоидозе доля плазматических клеток в костном мозге обычно не превышает 10%, костных поражений нет. В моче может присутствовать белок Бен-Джонса, а в сыворотке крови – небольшое количество парапротеина. Иногда первичный амилоидоз трансформируется в ММ.
4. Солитарная плазмоцитома может развиться как в кости, так и в мягких тканях. В сыворотке крови может присутствовать небольшое количество парапротеина. Иногда плазмоцитома может прогрессировать до ММ.
5. Плазмоклеточный лейкоз – тяжелое, быстро прогрессирующее заболевание, при котором в крови циркулирует большое количество плазматических клеток. Прогноз, как правило, неблагоприятный.
Современное лечение ММ включает применение цитостатических средств, глюкокортикоидов и анаболических гормонов, восстановительные методы, а также комплекс мероприятий, устраняющих или предупреждающих развитие метаболических нарушений и вторичного иммунодефицита.
Если заболевание диагностируют рано (I, частично II стадия болезни), то при отсутствии клинических симптомов, нормальных показателях крови (СОЭ не учитывают) и функций почек противоопухолевое лечение начинать не следует. Рекомендована выжидательная тактика с ежемесячным контролем показателей крови, мочи и секреции моноклонального парапротеина. У части таких больных существует «тлеющая» ММ, которая в течение нескольких лет не прогрессирует и не нуждается в лечении.
Лечение следует начинать при возникновении симптомов нарастания опухолевой массы (снижение концентрации гемоглобина и количества эритроцитов, повышение содержания парапротеина в крови или моче, сильные боли в костях).
При проведении цитостатической химиотерапии следует придерживаться определенных принципов.
1. Подбор цитостатического препарата осуществляют с учетом стадии болезни (величины опухолевой массы) и критериев риска.
2. Оценку эффективности лечения следует проводить на основании определенных признаков:
• снижение концентрации парапротеина в сыворотке крови более чем на 50%;
• снижение экскреции белка Бен-Джонса более чем на 50%;
• рентгенологические признаки заживления костных деструкций;
• уменьшение площади пораженных опухолью костей.
3. Требуется непрерывное лечение с соблюдением доз и интервалов в течение двух лет и более.
Применяют комбинацию цитостатического препарата мелфалана с преднизолоном. Существуют различные подходы к назначению этих лекарственных средств.
У больных с III стадией болезни при отсутствии явных признаков агрессивности (медленно прогрессирующая ММ) проводят пролонгированное лечение с поддерживающей терапией ударными прерывистыми курсами. Мелфалан сочетают с преднизолоном; одновременно назначают анаболические стероиды. Через 4 нед назначают поддерживающее лечение меньшими дозами используемых препаратов.
Еще один вариант пролонгированного лечения – применение винкристина в сочетании с мелфаланом и преднизолоном. Кроме того, возможно использование циклофосфана и преднизолона.
Другая методика – ударная прерывистая терапия – рекомендована больным с медленно прогрессирующей ММ I и II стадии. Применяют более короткие курсы лечения теми же препаратами – мелфаланом (циклофосфаном) в сочетании с преднизолоном.
При быстропрогрессирующей ММ с симптомами, указывающими на плохой прогноз, и резистентностью к ранее проводимому лечению назначают полихимиотерапию. В течение 3–4 нед назначают комбинацию винкристина, циклофосфана, мелфалана и преднизолона.
У молодых больных с резистентностью к лечению и отсутствием серьезных соматических заболеваний применяют так называемую интенсивную терапию.
Она включает использование высоких доз мелфалана в сочетании с трансплантацией костного мозга и тотальным облучением.
Для лечения ММ также применяют интерферон альфа, который не имеет самостоятельного значения, но рекомендовано его использование одновременно с химиотерапией, а также в перерывах между курсами. Интерферон альфа подавляет пролиферацию клона опухолевых клеток.
Лечение считают эффективным только у тех больных, у которых обнаруживают стабильность или улучшение показателей красной крови и содержания сывороточного альбумина, а также отсутствие увеличения размеров остеодеструктивных очагов. Эти критерии чрезвычайно важны, так как ориентация на степень снижения концентрации парапротеина не всегда верна: прямая зависимость между опухолевой массой и уровнем секреции парапротеина может быть весьма различной. Эффективность лечения оценивают через 3 мес после его начала. При отсутствии признаков улучшения больных относят к прогностически весьма неблагоприятным или так называемым нереагирующим.
Назначение локальной лучевой терапии рекомендовано во всех случаях угрозы патологических переломов позвоночника, крестцово-подвздошной области, бедренных и берцовых костей, даже при отсутствии болевого синдрома. Локальное облучение применяют при ограниченных опухолевых узлах в костях и мягких тканях, а также радикулярных болях, связанных со сдавлением корешков спинного мозга опухолью. Сочетать лучевое лечение и химиотерапию не рекомендуют.
На ранних стадиях ММ некоторым больным рекомендуют трансплантацию донорских стволовых клеток (с обычными условиями).
При инфекционных осложнениях следует применять антибиотики, не обладающие нефротоксичностью. При выраженной протеинемии и парапротеинемии проводят плазмаферез. При поражении костной ткани (переломы и др.) необходимо применять комплекс средств, улучшающих костную репарацию (внутримышечно миокальцик* э или прием внутрь препаратов кальция). Бифосфонаты весьма эффективны при лечении поражений костей, сопровождающих ММ, и могут увеличить продолжительность жизни пациентов. При переломах костей проводят иммобилизацию и вытяжение на щите (особенно при компрессионных переломах позвоночника).
Больные с I стадией ММ могут жить многие годы без какого-либо лечения. При развитии III стадии заболевания средняя продолжительность жизни больного составляет 2–3 года. Современное комбинированное лечение увеличивает ее: удается восстановить активность пациентов и поддерживать их удовлетворительное состояние. Больные погибают вследствие развития ХПН или инфекционных осложнений.
Анемия – состояние, характеризующееся уменьшением содержания гемоглобина в единице объема крови, вследствие снижения его общей концентрации в организме. В большинстве случаев анемия сопровождается снижением количества эритроцитов в единице объема крови. От истинной анемии следует отличать гидремию – разжижение крови за счет тканевой жидкости.
Необходимо отметить, что на сегодняшний день единой общепризнанной классификации анемий не существует.
Поскольку в основе развития анемий лежат различные патологические процессы, с патогенетической точки зрения предложено разделять все анемии на следующие группы.
1. Анемии, обусловленные нарушением синтеза гемоглобина:
• железодефицитная анемия (нарушение синтеза гема);
• сидероахрестическая анемия (нарушение синтеза порфирина);
• анемия хронических заболеваний.
2. Анемии вследствие нарушения образования и созревания эритроцитов (дисэритропоэтические анемии):
3. Анемии, обусловленные нарушениями пролиферации клеток костного мозга (гипопролиферативные анемии):
• идиопатическая гипопластическая (апластическая) анемия;
• вторичная гипопластическая (апластическая) анемия (вследствие действия лекарственных средств, токсинов, ионизирующей радиации и др.);
• миелофиброз (первичный или вторичный);
• замещение клеток костного мозга опухолевыми клетками (миелофтизис);
4. Анемии вследствие усиленного разрушения эритроцитов (гемолитические анемии):
• аутоиммунные гемолитические анемии;
• наследственная микросфероцитарная гемолитическая анемия (болезнь Минковского-Шоффара);
• пароксизмальная ночная гемоглобинурия.
5. Анемии вследствие дефицита эритропоэтина.
6. Анемии со смешанным механизмом развития.
Каждый из указанных патогенетических вариантов анемических состояний имеет различную этиологию (например, железодефицитная анемия может возникать при мено-, метроррагиях, кровотечениях из ЖКТ, при беременности, нарушении всасывания железа и др.). В ряде случаев самый тщательный диагностический поиск не позволяет обнаружить лежащее в основе анемии заболевание. В этом случае следует говорить об идиопатической форме анемии.
Именно поэтому при обследовании больного с предполагаемой анемией необходимо:
1. определить патогенетический вариант анемии;
2. определить заболевание, лежащее в основе анемии.
Симптомы анемий чрезвычайно разнообразны и определяются:
1. патогенетическим вариантом анемии;
3. изменениями в организме, которые обусловлены его реакцией на гипоксию тканей, вызванной нарушением дыхательной функции крови (доставка кислорода тканям), – циркуляторно-гипоксическим синдромом.
Циркуляторно-гипоксический синдром манифестирует слабостью, повышенной утомляемостью, одышкой при физической нагрузке, сердцебиениями, «анемическим» шумом в крупных сосудах, увеличением ОЦК и ускорением кровотока. В большей или меньшей степени он выражен при всех видах анемических состояний, а степень его выраженности зависит от уровня гипоксии тканей, что, в свою очередь, определяется кислородной емкостью крови.
Согласно классификации ВОЗ, в зависимости от тяжести выделяют:
1. анемии легкой степени тяжести (концентрация гемоглобина не ниже 90 г/л);
2. анемии средней степени тяжести (концентрация гемоглобина в пределах 90–70 г/л);
3. анемии выраженной степени тяжести или тяжелые (концентрация гемоглобина менее 70 г/л).
Помимо этого анемии классифицируют в зависимости от средних размеров эритроцитов и степени их насыщения гемоглобином. Последнюю определяют с помощью цветового показателя и по средней концентрации гемоглобина в эритроцитах (Mean Corpuscular Hemoglobin – МСН).
Цветовой показатель отражает степень насыщения эритроцитов гемоглобином. Его рассчитывают, умножив концентрацию гемоглобина (г/л) на 3 и разделив на первые две цифры числа эритроцитов.
В норме величина цветового показателя колеблется от 0,8 до 1,1.
В зависимости от насыщения эритроцитов гемоглобином все анемии можно разделить:
1. на нормохромные (цветовой показатель от 0,8 до 1,1 либо МСН от 30 до 36 г/дл);
2. гипохромные (цветовой показатель менее 0,8 либо МСН ниже 30 г/дл);
3. гиперхромные (цветовой показатель выше 1,1 либо МСН превышает 36 г/дл).
В зависимости от величины среднего объема эритроцитов, все анемии можно разделить:
1. на нормоцитарные (средний объем эритроцитов от 80 до 100 мкм 3 );
2. микроцитарные (средний объем эритроцитов менее 80 мкм 3 );
3. макроцитарные (средний объем эритроцитов более 100 мкм 3 ).
Сущность железодефицитной анемии (ЖДА) состоит в недостатке железа в организме (истощение его запасов в органах-депо), вследствие чего нарушается синтез гемоглобина, и каждый эритроцит содержит меньшее, чем в норме, количество гемоглобина. ЖДА регистрируют чаще остальных форм анемий, что можно объяснить множеством обстоятельств, приводящих к дефициту железа в организме.
Существует несколько основных причин дефицита железа.
1. Скрытые (оккультные) кровотечения:
• желудочно-кишечные (язвенная болезнь, геморрой, рак, диафрагмальная грыжа, НЯК, полипоз желудка и кишечника);
• маточные (дисфункция яичников, фибромиома матки, рак шейки матки, эндометриоз и др.);
• легочные (рак, бронхоэктазы, изолированный легочный гемосидероз).
2. Недостаточное потребление железа с пищей.
3. Повышенный расход железа:
• период роста и полового созревания;
• хронические инфекционные заболевания, опухоли.
4. Нарушение всасывания железа:
5. Нарушение транспорта железа (дефицит белка плазмы крови трансферрина).
Из перечисленных причин следует, что ЖДА чаще развивается у женщин в результате обильных маточных кровотечений и повторных беременностей, а также у подростков.
Важнейшая функция железа в организме – его участие в синтезе гема, служащего составной частью гемоглобина. При дефиците железа прежде всего возникает нарушение синтеза гемоглобина, что приводит к развитию ЖДА. Недостаточное образование гемоглобина служит причиной гипоксии тканей и развития циркуляторно-гипоксического синдрома. Дефицит железа также способствует нарушению синтеза тканевых ферментов, что приводит к изменению тканевого метаболизма, при этом происходит поражение быстро обновляющихся эпителиальных тканей – слизистой оболочки ЖКТ, кожи и ее дериватов. Патогенез ЖДА представлен на рис. 5–2.
Рис. 5–2. Патогенез железодефицитной анемии
В организме здорового взрослого человека общее количество железа составляет 3–4 г, при этом у женщин оно несколько меньше, чем у мужчин, что связано с ежемесячными потерями крови во время менструаций. При нормальном питании с пищей поступает 10–20 мг железа в сутки, но только 10% всасывается в двенадцатиперстной кишке и в верхних отделах тонкой кишки. Примерно такое же количество железа ежедневно образуется вследствие физиологического гемолиза эритроцитов.
Наибольшее количество железа содержится в чечевице, желтке куриного яйца и мясных продуктах (говядина).
Клиническая картина болезни, как это вытекает из схемы патогенеза, складывается из следующих синдромов:
1. циркуляторно-гипоксического (при достаточной выраженности анемии и кислородного голодания тканей);
2. поражения эпителиальных тканей (гастроэнтерологические расстройства, трофические нарушения кожи и ее дериватов);
3. гематологического (анемия гипохромного типа и признаки дефицита железа).
Кроме этих синдромов, клиническую картину также определяет заболевание, на основе которого развилась ЖДА (например, язвенная болезнь желудка или двенадцатиперстной кишки с повторными кровотечениями, мено- и метроррагии, какие-либо хронические инфекционные поражения и др.).
Имеет значение и стадия течения анемии:
1. скрытый дефицит железа, манифестирующий снижением концентрации сывороточного железа при отсутствии уменьшения содержания гемоглобина;
2. тканевый сидеропенический синдром (манифестирует гастроэнтерологическими расстройствами, трофическими изменениями кожи и ее дериватов);
3. анемия (снижение концентрации гемоглобина).
На первом этапе диагностического поиска при достаточно выраженной анемии можно обнаружить жалобы на слабость, шум в ушах, сердцебиение, одышку при физической нагрузке и ноющие боли в области сердца (признаки циркуляторно-гипоксического синдрома). Очень своеобразны гастроэнтерологические расстройства, представленные извращением вкуса и обоняния, снижением и извращением аппетита (желание есть мел, сухие макароны, зубной порошок), затруднением при глотании и неопределенными болевыми ощущениями в эпигастральной области. Нередко больные отмечают повышение температуры до субфебрильных цифр.
При умеренно выраженной анемии и дефиците железа все указанные жалобы могут быть выражены незначительно или отсутствовать. В анамнезе у таких больных есть указания на случайно обнаруженное снижение концентрации гемоглобина (например, во время профилактического осмотра). Пациенты могут предъявлять разнообразные жалобы, а также сообщать те или иные сведения о фоновом заболевании (состоянии), обусловившем возникновение дефицита железа и последующей анемии.
На втором этапе диагностического поиска следует активно искать симптомы поражения эпителиальной ткани и трофических расстройств кожи и ее дериватов (волос, ногтей). Так, можно обнаружить сглаженность сосочков языка, сухость и шелушение кожного покрова, ломкость ногтей, сухость и выпадение волос. Циркуляторно-гипоксический синдром манифестирует тахикардией, систолическим шумом над верхушкой сердца и на крупных сосудах (тоны сердца не изменены). На яремных венах можно выслушать шум «волчка». Кожный покров и слизистые оболочки обычно бледные; размеры селезенки, как правило, соответствуют норме. Ее умеренное увеличение обычно отмечают у тех больных, которым проводили многочисленные гемотрансфузии.
На третьем этапе диагностического поиска проводят исследования, результаты которых подтверждают не только существование и выраженность анемии, но и ее патогенетический вариант (обусловленность дефицитом железа).
При исследовании периферической крови обнаруживают снижение концентрации гемоглобина, микроцитоз (увеличение количества эритроцитов малого диаметра) и гипохромию эритроцитов, снижение цветового показателя и среднего содержания гемоглобина в эритроците (массовое и процентное). Содержание ретикулоцитов в норме или повышено. Изменяются показатели обмена железа: уменьшается содержание свободного железа в сыворотке крови и насыщение трансферрина железом; повышается общая железосвязывающая способность сыворотки и концентрация общего трансферрина. Это связано с тем, что в организме снижено содержание железа. Для определения его резервов железа в организме применяют десфераловую пробу. В норме взрослый человек после введения дефероксамина в дозе 500 мг теряет 0,6–1,3 мг железа с мочой. При ЖДА содержание железа в моче после введения препарата значительно ниже (0,2–0,4 мг), что указывает на уменьшение запасов железа в организме. Дефероксамин – продукт метаболизма актиномицетов, способный связывать железо. Известное представление об уменьшении запасов железа в организме можно получить, изучая всасывание радиоактивного железа. При ЖДА оно повышается.
В костном мозге при ЖДА отмечают уменьшение количества сидеробластов – эритрокариоцитов, содержащих железо (как известно, в норме 20–40% эритрокариоцитов костного мозга содержат единичные гранулы железа). В ряде случаев гранулы обнаружить не удается.
При обследовании ЖКТ достаточно часто обнаруживают снижение желудочной секреции (базальной и стимулированной), а также атрофические изменения слизистой оболочки пищевода и желудка.
При выраженном циркуляторно-гипоксическом синдроме можно обнаружить признаки поражения миокарда (миокардиодистрофия вследствие анемии) в виде умеренного расширения сердца (определяют при рентгенологическом исследовании) и изменений конечной части ЭКГ (снижение амплитуды или отрицательный зубец Т, преимущественно в грудных отведениях).
Выделяют два этапа диагностики ЖДА:
1. сбор доказательств дефицита железа в организме, послужившего причиной анемии;
2. установление причин развития железодефицитного состояния.
Критерии дефицита железа и анемии:
1. концентрация гемоглобина ниже 120 г/л у мужчин и ниже 116 г/л у женщин;
2. снижение цветового показателя ниже 0,8;
3. снижение среднего содержания гемоглобина в эритроцитах (менее 24 пг);
4. снижение средней концентрации гемоглобина в эритроцитах (ниже 30%);
5. увеличение количества микроцитов (эритроцитов диаметром менее 6 мкм) более 20%;
6. снижение концентрации сывороточного железа менее 11,6 мкмоль/л;
7. повышение содержания свободного (более 35,8 мкмоль/л) и общего трансферрина (общей железосвязывающей способности сыворотки) более 71,6 мкмоль/л;
8. снижение насыщения трансферрина железом (менее 25%);
9. усиление всасывания радиоактивного железа;
10. положительная проба с дефероксамином (уменьшение содержания железа в моче после введения дефероксамина).
Для установления причины железодефицитного состояния прежде всего необходимо найти источник кровотечения. Для этого, наряду с тщательным клиническим обследованием, требуется проведение эндоскопических (эзофагогастродуоденоскопия, ректоромано- и колоноскопия, бронхоскопия) и других исследований. Женщин обязательно должен осмотреть гинеколог.
Обнаружить скрытые (оккультные) кровотечения очень трудно. Если не удалось установить их источник, то применяют пробу с введением больному его собственных эритроцитов, предварительно меченных 51 Сr, а в дальнейшем определяют радиоактивность кала. Ее повышение свидетельствует об источнике кровотечения в ЖКТ.
При хронических инфекционных заболеваниях большое значение имеет определение концентрации свободного трансферрина в крови (латентная железосвязывающая способность сыворотки), которая, в отличие от постгеморрагических анемий, остается нормальной.
ЖДА следует дифференцировать от сидероахрестической анемии и талассемии (один из видов наследственной гемолитической анемии). При сидероахрестической анемии вследствие генетического или приобретенного нарушения обмена порфиринов железо не поступает в эритроидные клетки. В результате этого развивается анемия с резким снижением цветового показателя при повышенном содержании железа в крови. В костном мозге – раздражение красного ростка и повышенное содержание эритроидных клеток с включением железа. Лечение его препаратами при сидероахрестической анемии безуспешно.
При талассемии (более подробно см. «Гемолитические анемии») отмечают умеренное снижение содержания гемоглобина при значительном уменьшении цветового показателя. Концентрация сывороточного железа повышена. Характерно обнаружение мишеневидных эритроцитов. Одновременно отмечают все признаки гемолитического синдрома.
Формулировка развернутого клинического диагноза ЖДА должна включать следующие компоненты:
1. определение характера анемии (в данном случае – железодефицитная);
2. указание этиологии заболевания;
3. определение стадии процесса (ремиссия или рецидив, который может характеризоваться скрытым дефицитом железа).
Воздействуют на этиологические факторы (удаление источника кровотечения, борьба с инфекционным поражением, противоопухолевое лечение, профилактика врожденного дефицита железа) и проводят патогенетическую терапию (ликвидация дефицита железа, борьба с серьезными расстройствами гемодинамики).
Рацион больных ЖДА должен включать продукты, богатые железом, но следует учитывать не только содержание железа в них, но и степень всасывания микроэлемента. Наибольшее количество железа содержат мясные продукты (говядина, телятина). Содержащееся в них гемовое железо всасывается на 25–30%. Всасывание железа из рыбы ниже (до 10%), из растительных продуктов – всего 3–5%. Таким образом, ликвидацию дефицита железа осуществляют с помощью приема внутрь или парентерального введения препаратов железа. Необходимо, чтобы суточная доза двухвалентного железа (только оно подвергается всасыванию) составляла 100–300 мг. В связи с этим при выборе препарата железа и определении его суточной дозы необходимо ориентироваться не только на общее содержание в нем железа, но и на концентрацию двухвалентного железа. Естественно, что предпочтительнее назначать средства с высоким содержанием последнего. Это связано с удобством их приема больными (1–2 раза в сутки). Входящие в состав многих лекарственных форм аскорбиновая и янтарная кислота, фруктоза и цистеин усиливают всасывание железа. Для улучшения последнего препараты железа следует принимать до приема пищи.
Основной принцип лечения препаратами железа – их длительное применение в достаточных дозах. Только в этом случае можно получить стойкий результат. Довольно давно применяют препарат железа сульфат в сочетании с аскорбиновой кислотой (по 15–20 драже в сутки). В настоящее время существуют препараты железа, содержащие двухвалентное железо в существенно большей концентрации, что позволяет обойтись приемом препарата 1–2 раза в день. Сорбифер дурулес* принимают по 1–2 таблетки в день, железа сульфат – по две таблетки в день. Представляет интерес новый препарат железа сульфат + фолиевая кислота + цианокобаламин, содержащий кроме сульфата железа аскорбиновую кислоту в дозе 100 мг, цианокобаламин в дозе 10 мкг и фолиевую кислоту в дозе 5 мг. Препарат можно принимать по 1–2 таблетки в сутки. Железа сульфат + серин – назначают по одной капсуле 2–3 раза в день или в виде сиропа (по одной чайной ложке на 12 кг массы тела). Сироп нельзя назначать больным сахарным диабетом, так как он содержит много сахара. В лекарственном средстве поливитамины + минералы железо содержится в микродиализных капсулах, что обеспечивает постоянство скорости его высвобождения (плазменной концентрации препарата) в течение суток. Для парентерального введения используют железа гидроксид полиизомальтозат и фербитол*. Железа гидроксид полиизомальтозат для внутримышечного введения выпускают в ампулах по 2 мл, содержащих 100 мг железа (соединение окиси трехвалентного железа с полиизомальтозой), а для внутривенного введения – в ампулах по 5 мл коллоидного раствора, в котором железо связано с натриево-сахаратным комплексом. Новый препарат для внутримышечного введения – железа гидроксид полимальтозат, для внутривенного введения – железа гидроксид сахарозный комплекс.
При назначении препаратов железа в достаточной дозе на 7–10-й день после начала лечения отмечают увеличение количества ретикулоцитов в периферической крови (ретикулоцитарный криз). Прирост концентрации гемоглобина начинается через 3–4 нед после его начала, но в ряде случаев это может произойти на 6–8-й нед. Лечение следует проводить не менее 3 мес. После достижения ремиссии больным с продолжающимися кровотечениями (например, при меноррагиях) следует рекомендовать поддерживающее лечение тем же препаратом (ежемесячно по 7–10 дней).
Ряду пациентов назначают парентеральное введение препаратов железа.
1. тошнота, рвота (непереносимость препаратов железа при приеме внутрь, что не позволяет продолжать дальнейшее лечение);
2. нарушение всасывания при патологических изменениях кишечника (энтериты, резекция тонкой кишки, синдром недостаточного всасывания);
3. нежелательность приема внутрь препаратов железа больными с заболеваниями ЖКТ (обострение язвенной болезни желудка или двенадцатиперстной кишки, БК, НЯК);
4. необходимость более быстрого насыщения организма железом (особенно в ситуациях, когда планируют оперативное вмешательство).
При аллергических реакциях на парентеральное введение препаратов железа и непереносимости их приема внутрь следует проводить трансфузии эритроцитной массы. Переливание крови способствует быстрому увеличению содержания гемоглобина, но его утилизация при этом значительно ограничена. Кроме того, существует опасность заражения больных инфекционным мононуклеозом, сывороточным гепатитом и др. В связи с этим гемотрансфузии проводят лишь по жизненным показаниям (при подготовке к оперативному вмешательству, выраженных гемодинамических нарушениях, связанных с анемией). В последнем случае следует стремиться не к нормализации концентрации гемоглобина путем гемотрансфузий, а к улучшению общего состояния больного.
Ликвидация причины потери крови, а также систематический прием препаратов железа приводят к полному выздоровлению. У женщин с обильными маточными кровотечениями необходим систематический контроль содержания гемоглобина (как правило, их ставят на диспансерный учет).
Лица, подверженные опасности развития дефицита железа (недоношенные дети, дети от многоплодной беременности, девушки в период полового созревания при быстром росте, женщины с обильными менструациями, беременные), должны употреблять продукты с достаточным содержанием железа (прежде всего, говядину). Им рекомендовано периодическое исследование крови для определения скрытого дефицита железа и анемии.
источник
Множественная миелома — это сложное заболевание, которое может вызвать множество симптомов. Между прочим, вы можете ощущать боль в костях, беспокойство, замешательство, усталость и потерю аппетита.
Эти симптомы могут заставлять вас говорить с врачом, что приводит к множественному диагнозу миеломы.
Люди с множественной миеломой испытывают усталость из-за низкого количества эритроцитов, вызванного раком. «Анемия» — это термин, используемый для описания низкого количества этих клеток.
Согласно Исследовательскому фонду множественных миелом (MMRF), около 60 процентов людей с множественной миеломой имеют анемию во время диагноза.
Анемия является результатом сокращения эритроцитов в организме. Существуют разные причины этого состояния. Некоторые люди развивают анемию, потому что у них есть заболевание, вызывающее кровотечение. Другие развивают его из-за состояния, которое вызывает снижение производства эритроцитов из их костного мозга.
Анемия и множественная миелома идут рука об руку. Множественная миелома вызывает разрастание плазматических клеток в костном мозге. Плазменные клетки представляют собой белые кровяные клетки, которые продуцируют и выделяют антитела. Слишком много из этих клеток в толпе костного мозга и уменьшают количество нормальных кроветворных клеток. Этот ответ вызывает низкий уровень эритроцитов.
Состояние может быть умеренным, умеренным, тяжелым или опасным для жизни. Эритроциты содержат гемоглобин. Гемоглобин переносит кислород из легких в разные части тела. Ваш врач может диагностировать анемию, если уровень гемоглобина ниже нормы. Для женщин нормальный уровень гемоглобина составляет от 12 до 16 граммов на децилитр (г / дл). Для мужчин нормальный уровень составляет от 14 до 18 г / дл.
Симптомы анемии могут включать:
- головокружение
- одышка
- головная боль
- холодность
- боль в груди
- бледная кожа
- низкая энергия > аритмия
- Какова связь между анемией и множественным лечением миеломы?
Анемия также может развиваться как побочный эффект некоторых методов лечения рака. Некоторые лекарства уменьшают количество эритроцитов, вырабатываемых организмом.
Поговорите со своим врачом, чтобы понять возможные осложнения различных методов лечения. Лечение рака, которое может вызвать низкий уровень крови, включают:
- Это лечение также может убивать здоровые клетки вместе со злокачественными клетками. Эти здоровые клетки включают клетки в костном мозге, которые образуют эритроциты. Излучение.
- Эта терапия использует высокоэнергетические рентгеновские лучи для сокращения опухолей и повреждения раковых клеток. Он может также повредить костный мозг, когда он проводится на больших участках тела (кости, сундук, живот или таз). Такой ущерб приводит к снижению производства эритроцитов. Анемия обычно является временной. Поскольку ваш рак улучшается, ваше производство красных кровяных телец должно нормализоваться.
Как лечить анемию с множественной миеломой
Анемия может вызвать множество симптомов, включая низкую энергию, головокружение, головные боли и повреждение органов. Ваш врач может предложить лечение, чтобы помочь восстановить нормальное количество эритроцитов во время полной терапии рака.
Ваш врач может контролировать количество клеток крови с помощью анализов крови. Это может выявить анемию, а также оценить эффективность конкретного лечения. Варианты лечения анемии варьируются, но могут включать:
Дефицит витамина может вызвать анемию при множественной миеломе. Ваш врач может заказать анализ крови, чтобы определить, есть ли у вас дефицит. Если вы это сделаете, они порекомендуют дополнение, чтобы исправить этот недостаток.
Витаминные добавки могут включать железо, фолиевую кислоту или витамин B-12. Ваш врач может рекомендовать внебиржевые добавки и диетические изменения. В зависимости от тяжести анемии, ваш врач может назначить дополнение или витамин B-12.
Лекарства также доступны для запуска вашего производства костного мозга эритроцитов. Это может привести к анемии и ее симптомам. Такие препараты включают эпоэтин альфа (Procrit или Epogren) и дарбепоэтин альфа (Aranesp).
Хотя они эффективны, эти лекарства не для всех. Существует риск сгустков крови в сочетании с некоторыми препаратами, которые лечат множественную миелому. Ваш врач может определить, безопасно ли принимать один из вышеуказанных препаратов с вашей текущей терапией.
Когда анемия тяжелая или опасная для жизни, ваш врач может рекомендовать переливание крови.
Поговорите со своим врачом, как только вы обнаружите признаки анемии. Вам может понадобиться добавка витамина, чтобы увеличить ваше производство красных кровяных телец. Или вы также можете быть кандидатом на лечение.
Анемия может улучшиться по мере того, как вы достигнете ремиссии, и ваш костный мозг станет более здоровым.
источник