Наследственные гемолитические анемии — результат врожденного дефекта гемоглобина, ферментов или мембран эритроцитов. Соответственно выделяют три группы наследственных гемолитических анемий:
- Гемоглобинопатии (серповидноклеточная анемия, гемоглобинопатия HbSC).
- Ферментопатии (анемии при недостаточности глюкозо-6-фосфат дегидрогеназы).
- Мембранопатии (наследственный микросфероцитоз, овалоцитоз и пойкилоцитоз).
Серповидноклеточная анемия — наследственное заболевание системы крови, характеризующееся генетическим дефектом, в результате которого происходит нарушение строения белка гемоглобина (гемоглобинопатия). Образующийся при этом аномальный гемоглобин S (HbS) отличается по своим электрофизиологическим свойствам от нормального гемоглобина (HbA) здорового человека, в результате чего изменяются и сами эритроциты, приобретая характерную удлиненную форму, под микроскопом напоминающую серп (отсюда и название заболевания).
Серповидноклеточная анемия — заболевание, развивающееся у гомозигот по аллелю, кодирующему гемоглобин S, имеющий структурные аномалии молекулы гемоглобина и способный к полимеризации при отдаче кислорода или снижении его парциального давления. В эритроцитах образуются волокна (тактоиды), формирующие студенистую сеть, изменяющие форму эритроцитов на серповидную и повышающие их жесткость, что затрудняет прохождение эритроцитов по мелким сосудам. Как следствие возникает закупорка этих сосудов, и развиваются многочисленные инфаркты в селезенке, легких, почках и головном мозге.
Заболевание проявляется обычно в детском возрасте. Характерны бледность, утомляемость, задержка роста, повышенная чувствительность к инфекциям. В результате хронического гемолиза развиваются желтуха и желчнокаменная болезнь.
Характерными симптомами анемии являются трофические язвы голеней, приапизм, повторные инфаркты легких, хроническое легочное сердце, некроз почечных сосочков, обусловленные ухудшением реологических свойств крови. Возможно развитие кровоизлияний в сетчатку с последующими рубцеванием и отслойкой сетчатки.
Заболевание является хроническим. Течение заболевания нередко осложняется инфекцией, в частности сальмонеллезом, приводящим к остеомиелиту. Иногда возникает асептический некроз головки бедренной кости. Тяжелым осложнением и у детей, и у взрослых является инсульт.
Нередко развиваются угрожающие жизни больных острые кризы. Самый частый из них — болевой криз. Боль локализуется в спине, конечностях и ребрах, длится несколько дней или недель, сопровождается лихорадкой, но концентрация гемоглобина в крови нормальная. Возможно развитие острого синдрома грудной клетки, проявляющегося болью в грудной клетке, легочными инфильтратами и гипоксией.
У детей, а иногда и у взрослых, со спленомегалией развиваются секвестрационные кризы, характеризующиеся депонированием крови в селезенке и сопровождающиеся артериальной гипотонией и шоком, падением концентрации гемоглобина в крови.
Редко развиваются гемолитические кризы, характеризующиеся вторичной желтухой и снижением концентрации гемоглобина в крови.
При вирусной инфекции (обычно парвавирусе В19) развиваются апластические кризы, сопровождающиеся резким снижением гемоглобина и уменьшением количества ретикулоцитов в периферической крови.
Сходные клинические проявления имеет гемоглобинопатия HbSC. Она протекает менее тяжело, но с выраженной спленомегалией.
Гемоглобин S при серповидноклеточной анемии выявляют после обработки эритроцитов метабисульфатом натрия, что приводит к отдаче кислорода. С помощью электрофореза (более точный метод) можно количественно определить гемоглобин S и отличить гомозиготную от гетерозиготной анемии или от других аномалий строения гемоглобина.
Гемоглобин при серповидноклеточной анемии снижен до 50–100 г/л, при гетерозиготности по HbS его уровень нормальный. Средний эритроцитарный объем может быть увеличенным, наблюдается непрямая гипербилирубинемия и нейтрофильный лейкоцитоз, количество тромбоцитов повышено.
В мазках периферической крови выявляют серповидные эритроциты, иногда тельца Говела–Жолли и мишеневидные эритроциты.
Специфическим методом лечения является лечение гидроксимочевиной, повышающей уровень фетального гемоглобина и снижающей уровень гемолиза. Эффективность препарата усиливается назначением эритропоэтина. Этот метод лечения применяют только больным с тяжелой анемией.
Основное лечение направлено на предупреждение острых и хронических осложнений. Не следует допускать обезвоживания, длительного пребывания на большой высоте.
Детям от 3-х месяцев до 5 лет показано введение менингококковой вакцины и вакцины против Haemophilus influenza типа В. Детям старше 5 лет назначают профилактическую терапию пенициллином по 125–250 мг внутрь ежедневно. В случае лихорадки срочно проводят интенсивную противомикробную терапию.
Всем больным с хронической гемолитической анемией назначают фолиевую кислоту 1 мг внутрь 1 раз в сутки.
При болевом кризе в/в вводят жидкость и проводят обезболивающую терапию анальгетиками. Наиболее эффективны длительные инфузии морфина. Но следует помнить о склонности больных с рецидивирующим болевым синдромом к наркотической зависимости. Переливание крови не устраняет боль, повторные же переливания могут привести к гемосидерозу.
При остром синдроме грудной клетки проводят ингаляции кислорода и инфузионную терапию и назначают антибиотики широкого спектра действия (цефтриаксон и эритромицин). При снижении РаО2 в крови ниже 60 мм рт. ст. проводят обменное переливание крови.
Лечение секвестрационного криза направлено на восстановление гемодинамики. Основным лечением апластического криза является трансфузионная терапия.
Лечение остеомиелита проводят с учетом результата бактериологического исследования биопсийного материала. Трофические язвы голени лечат возвышенным положением нижних конечностей, интенсивными местными воздействиями и при отсутствии эффекта проводят трансфузионную терапию и пересадки полнослойных лоскутов кожи. Приапизм устраняется регидратационной терапией или хирургической декомпрессией.
Излечение от серповидноклеточной анемии у некоторых больных возможно с помощью аллотрансплантации костного мозга. Но отбор для этого лечения сложен и широко не применяется. В настоящее время интенсивно развиваются методы генотерапии.
Ферментопатические гемолитические анемии — группа заболеваний, обусловленных дефицитом активности эритроцитарных ферментов. Врожденный дефицит ферментов (пируваткиназы, глюкозо-6-фосфатизомеразы, гексокиназы, глюкозо-6-фосфатдегидрогеназы) приводит к развитию анемии. Чаще всего встречается дефицит глюкозо-6-фосфатдегидрогеназы.
Нормальные эритроциты защищены от действия окислителей за счет метаболизма глюкозы по пентозофосфатному пути, являющемуся источником восстановленного глутатиона, препятствующего окислению сульфгидрильных групп гемоглобина и мембраны эритроцитов. Образующиеся свободные радикалы кислорода при контакте с токсинами или лекарствами в эритроцитах резко увеличивают метаболизм глюкозы по этому пути.
При недостаточности глюкозо-6-фосфатдегидрогеназы в эритроцитах не образуется необходимое количество восстановленного глутатиона, что приводит к окислению сульфгидрильнх групп гемоглобина с образованием телец Гейнца и мембран эритроцитов и развитию хронического или эпизодического гемолиза.
Гем глюкозо-6-фосфатдегидрогеназы расположен на Х-хромосоме. Поэтому недостаточность глюкозо-6-фосфатдегидрогеназы наследуется сцеплено с Х-хромосомой главным образом у мужчин — выходцев из Африки, Средиземноморья и Китая. У женщин заболевание наблюдается редко.
Гемолитические кризы — основное клиническое проявление этой ферментопатической анемии — развиваются обычно быстро, в течение нескольких часов и только под действием провоцирующих факторов. К ним относятся лекарственные средства (сульфаниламиды, хинин, нитрофурантоин, аспирин, феназопиридин), инфекции и бобы, принимаемые с пищей.
Содержание глюкозо-6-фосфатдегидрогеназы снижено главным образом в стареющих эритроцитах, и они более подвержены гемолизу. В момент криза в мазках периферической крови выявляются тельца Гейнца. Характерно быстрое снижение гематокрита, повышение в плазме концентрации свободного гемоглобина и непрямого билирубина и снижение уровня гаптоглобина. Гемолитический криз проходит быстро, обычно самостоятельно после разрушения старых эритроцитов.
К концу первых суток тельца Гейнца удаляются селезенкой. После их удаления в крови появляются эритроциты с полукруглыми краевыми дефектами («укусы» эритроцитов), фрагменты эритроцитов и небольшое количество микросфероцитов. После криза уровень глюкозо-6-фосфатдегидрогеназы может нормализоваться за счет преобладания в крови популяции молодых эритроцитов. Активность фермента следует определить через 1,5–2 мес после криза.
Основное внимание следует уделить профилактике гемолитических кризов: лечению инфекции, исключению приема некоторых лекарственных средств и употребления в пищу бобов. Гемотрансфузии проводят по строгим показателям. Во время кризов с гемоглобинурией проводят адекватную гидратацию для поддержания нормальной функции почек.
Мембранопатии — наследственные заболевания, характеризующиеся структурными аномалиями белков эритроцитов, приводящими к нарушениям структуры их мембран и внесосудистому гемолизу. Различают три формы наследственных мембранопатий: микросфероцитоз, овалоцитоз и стоматоцитоз.
Заболевание наследуется в большинстве случаев по аутосомно-доминантному типу и характеризуется дефектом одного из белков цитоскелета эритроцитов, приводящим к потере части их мембраны с уменьшением отношения площади к объему. Эритроциты превращаются в микросфероциты, имеющие средний диаметр меньше 6,3 мкм и среднюю толщину больше 2,1 мкм.
Наследственный микросфероцитоз может выявиться как у детей, так и у взрослых. У больных находят синдром анемии, желтуху и спленомегалию. Желтуха сопровождается образованием пигментных камней в желчном пузыре и желчевыводящих путях даже у детей. В костном мозге развивается гиперплазия эритроидного ростка.
Анемия нормохромная, умеренная (гемоглобин 90–100 г/л). В мазке крови выявляют микросфероциты — мелкие клетки без центрального просветления. Характерна способность микросфероцитов к гемолизу в гипотонических растворах меньшей концентрации в отличие от нормальных эритроцитов. Секвестрация микросфероцитов происходит в селезенке.
Диагностическое значение имеет спонтанный гемолиз — число эритроцитов, разрушившихся после инкубации крови в течение 48 ч. У здоровых людей он не превышает 4%, у больных с наследственным микросфероцитозом достигает 15–50%. Спонтанный гемолиз при добавлении в кровь глюкозы резко снижается. Прямая проба Кумбса отрицательная в отличие от аутоиммунной гемолитической анемии, при которой она положительна.
Клиническое выздоровление наступает после спленэктомии, хотя микроцитоз остается, но гемолиз значительно уменьшается. С профилактической целью назначают длительно фолиевую кислоту (1 мг/сут внутрь). При наличии желчнокаменной болезни после спленэктомии решается вопрос о холецистэктомии.
Наследственный овалоцитоз — заболевание, сопровождающееся появлением в крови большого количества овалоцитов. В периферической крови здоровых лиц количество их составляет 5–10%, а у больных колеблется от 25 до 90%. Овалоцитоз наследуется по аутосомно-доминантному типу.
Ведущее значение в патогенезе заболевания имеет структурная аномалия спектрина, приводящая к образованию измененного цитоскелета эритроцитов. Возможна недостаточность белка 4,1 цитоскелета, связывающего спектрин и актин. В результате имеющие двояковогнутую форму диски эритроцитов после прохождения микроциркуляторного русла становятся овальными и не восстанавливают свою прежнюю форму. Овалоциты разрушаются главным образом в селезенке.
У большинства больных овалоцитоз обнаруживается случайно при исследовании периферической крови или проявляется легким гемолизом с нормальной концентрацией гемоглобина (больше 120 г/л). Концентрация гемоглобина может быть сниженной. У 10–15% больных выявляется тяжелое течение заболевания, характеризующееся интенсивным гемолизом. Концентрация гемоглобина снижается до 90–100 г/л и ниже, продолжительность жизни половины эритроцитов составляет 5 сут. В периферической крови обнаруживают микроовалоциты, пойкилоциты и шизоциты.
Лечение наследственного овалоцитоза аналогично лечению наследственного микросфероцитоза. Основное значение имеет спленэктомия, устраняющая гемолиз.
Наследственный стомацитоз — заболевание, при котором эритроциты имеют специфическую форму — выпуклые с одной стороны и вогнутые с другой и имеют щелевидное пространство в окрашенном мазке крови.
Существует две формы эритроцитов:
- гипергидратированные стоматоциты (гидроциты) с высоким содержанием ионов натрия и воды и низкой средней концентрацией гемоглобина;
- дегидратированные стоматоциты (ксероциты) с низким содержанием ионов натрия и воды, и высокой средней концентрацией гемоглобина.
Гидроциты выглядят в окрашенных мазках крови как типичные стоматоциты, а ксероциты сморщенные и имеют вид мишеней. Стоматоциты имеют укороченную продолжительность жизни.
Клиника наследственного стоматоцитоза проявляется спленомегалией и легким гемолизом. Спленэктомия полностью гемолиз не устраняет.
источник
К наследственным гемолитическим анемиям относятся:
Гемоглобинопатии — группа наследственных гемолитических анемий, происхождение которых связано с нарушением синтеза или структуры полипептидных цепей гемоглобина. Нормальный гемоглобин — Hb A1 — характеризуется наличием 4- цепей: (α1 ,α2,
b1, b2 ). Качественные изменения гемоглобина характеризуются нарушением его структуры. Если в b2-цепи в положении b6 глутамат заменяется на валин, то развивается серповидноклеточная анемия с образованием HbS. При снижении напряжения кислорода в артериальной крови этот гемоглобин выпадает в кристаллы и вызывает деформацию эритроцитов. Тип наследования — рецессивный. Если в положении b6 глутамат заменяется на лизин, то образуется Hb C, развивается мишеневидноклеточная анемия. Тип наследования этой анемии — аутосомно-рецессивный.
В ряде случаев структура гемоглобина может не нарушаться, но изменяется скорость синтеза цепей гемоглобина. Это — количественные гемоглобинопатии, талассемии. Если изменяется скорость синтеза α-цепи, то развивается α-талассемия. Если изменяется скорость синтеза b -цепи — b-талассемия. Чаще развивается b — талассемия. Тип наследования — аутосомно-доминантный. Гомозиготы быстро погибают.
Мембранопатии — группа наследственных гемолитических анемий, связанная с аномалией белковых или липидных компонентов мембран эритроцитов, что является причиной изменения их формы и преждевременного их разрушения. Эти анемии связаны с генетическим дефицитом в мембране эритроцитов Ca 2+ -зависимой АТФазы, холестерина и фосфолипидов. Тип наследования — аутосомно-доминантный. При мембранопатиях повышается проницаемость мембраны эритроцитов под влиянием свободных радикалов, пероксидов. В результате в эритроцит поступает натрий, который связывает воду. Эритроцит набухает, становится сферическим и быстро разрушается. Осмотическая резистетнтность его снижена. К мембранопатиям относится сфероцитарная анемия Минковского -Шоффара.
Na + Na + Гемолиз эритроцитов
Эритроэнзимопатии — гемолитические анемии, возникающие в результате наследственной недостаточности ферментов в мембране эритроцитов. Примером эритроэнзимопатий служит наследственный дефицит фермента глюкозо-6-фосфатдегидрогеназы (Гл-6-ФДГ), наследуемый по доминантному типу, сцепленному с полом. Так, при недостатке фермента Гл-6-ФДГ блокируется реакция окисления глюкозо-6-фосфата в пентозофосфатном цикле, вследствие чего уменьшается образование восстановленной формы глутатиона, предохраняющего сульфгидрильные группы мембраны эритроцитов от действия различных окислителей (пероксидов липидов, фтивазида, свободных радикалов, сульфаниламидов). Гемолиз эритроцитов при дефиците Гл-6-ФДГ может возникнуть при употреблении в пищу бобов растительного происхождения, конских бобов (Vicia fava). Развивается заболевание фавизм. Болеют чаще дети в возрасте от 1 года до 14 лет., преимущественно мальчики-гемизиготы (одинарный набор хромосом), носители патологической X-хромосомы. Болеют также девочки-гомозиготы с аномальной X-хромосомой
Гл-6-ФДГ ПФЦ — Гл-6-Фосфат глутатион Эритроцит
Нарушение переваривания и всасывания жиров. Механизмы развития. Недостаточное и избыточное поступление жира в организм, последствия.
Нарушение переваривания
Расщепление жиров в кишечнике происходит при участии панкреатической липазы. Секреция липазы и ее активность зависит от активности дигестивных гормонов (холецистокинина, секретина), которые вырабатываются в слизистой тонкой кишки. При воспалительных процессах желудочно-кишечного тракта выработка этих гормонов нарушается, что влияет на характер переваривания жиров. Секретин определяет количество выделяемого сока поджелудочной железой и липазы. Качество панкреатического сока и активность липазы определяется холецистокинином. Причинами нарушения выработки панкреатического сока и липазы являются воспалительные процессы в поджелудочной железе, сдавление и спазм протоков, камни в протоках. Важную роль в переваривании жиров играет желчь. Желчь эмульгирует жиры и они легче поддаются действию липазы. Холецитокинин способствует выходу желчи из печени в кишечник. Нарушение желчевыделения может быть связано с воспалительными процессами в печени и желчных путях, дискинезиями, желчнокаменной болезнью. Проявлениями нарушений процессов переваривания являются болевой синдром и развитие стеаторреи — жирного поноса. В норме из организма выводится около 10% жиров, при нарушении переваривания — до 50%.
Нарушение всасывания
Для нормального всасывания жиры должны связываться с желчными кислотами и образовывать мицеллы. Дальнейшее всасывание жирных кислот происходит с участием энтероцитов, которые извлекают жирные кислоты из мицелл. Около 5% жирных кислот поступает в кровь путем простой диффузии. Основная масса жиров ресинтезируется с образованием триглицеридов. В крови жирные кислоты связываются с белками (альбуминами) и образуют липопротеиновые комплексы. Основным местом их образования является печень. Нарушение всасывания жирных кислот наблюдается при воспалении желудочно-кишечного тракта (энтериты), дистрофических процессах в слизистой кишечника, при увеличении содержания ионов кальция, связывающего жирные кислоты и затрудняющего поступления их из кишечника в кровь, при гиповитаминозе А и С. Нарушение всасывания жирных кислот приводит к гиполипемии — снижению содержания липидов в крови. Развивается гипоэргоз, нарушается всасывание жирорастворимых витаминов — А, Д, К, Е, развивается полигиповитаминоз. Нарушение всасывания липидов сопровождается диспепсией.
нарушения жирового обмена проявляется в виде:
Избыточное отложение жира в жировой ткани занимает ведущее место среди других нарушений обмена веществ. Среди взрослого населения от 30% до60% лиц имеет избыточный вес.
По этиологии выделяют ожирение трех видов: церебральное (16-20% случаев), алиментарное (55-66%), гормональное (около 20%).
По характеру накопления жира различают гиперпластическое ожирение, характеризующееся увеличением количества жировых клеток, и гипертрофическое, связанное с увеличением объема жировых клеток.
Различают 4 степени ожирения:
1 степень — увеличение веса на 30%
2 степень — увеличение веса на 50%
3 степень — увеличение веса на 100%
4 степень — увеличение веса на 200%
2. Метаболическое ожирение
Алиментарное ожирение возникает при переедании, гиподинамии. В основе его развития лежит повышение реактивности периферических и центральных рецепторов. Повышается порог возбудимости рецепторов желудочно-кишечного тракта, что приводит к изменению реактивности центральных рецепторов. Повышается тонус пищевого центра, в частности, вентролатерального ядра гипоталамуса (центр голода). С другой стороны, снижается возбудимость центра сытости (вентромедиальное ядро гипоталамуса).
В основе этого механизма лежат нейрогормональные механизмы.
Основные виды метаболического ожирения
1. Церебральное (гипоталамическое)
Ожирение при участии нейрогормональных механизмов обусловлено избыточным образованием жира или задержкой его в жировых депо.
Избыточное образование жира
Избыточное образование жира связано с активацией пентозо-фосфатного цикла (ПФЦ) и возрастанием активности фермента никотинамиддинуклеотид фосфат восстановленный (НАДФ.Н). Активация ПФЦ может быть наследственного происхождения, при гиперсекреции инсулина, образовании жира из аминокислот.
Задержка жира в жировых депо обусловлена снижением активности фермента липазы, нарушением расщепления жиров и замедлением поступления липидов в кровь. Угнетение активности липазы может возникать при снижении тонуса симпатической нервной системы и уменьшении выработки адреналина, при гиперсекреции инсулина.
Исхудание обусловлено уменьшением поступления и всасывания жиров (голодание, воспалительные процессы желудочно-кишечного тракта) и нарушением отложения жиров в депо, при нарушении нейрогормональной регуляции жирового обмена, связанное с повышением активности симпатической нервной системы, при стрессе.
2. Ведущие звенья патогенеза: нейрогуморальные, метаболические, иммунные и генетические механизмы заболеваний. Местные и общие реакции на повреждения, первичные и вторичные повреждения, их взаимосвязь; «порочные круги» в патогенезе.
Pathos — болезнь, genesis — развитие
Патогенез — это наука о механизмах развития заболевания. Существуют общие механизмы патогенеза:
3. Метаболические механизмы
Нейрогенные механизмы
Нейрогенные механизмы включают: нейрорецепторы, афферентное, центральное и эфферентное звенья.
1. Нейрорецепторы являются первичным звеном в этом механизме при действии чрезвычайного раздражителя. Его действие на нейрорецепторы происходит опосредованно, рефлекторно. Кроме нейрорецепторов, есть клеточные рецепторы, раздражение которых обусловлено прямым действием патогенного фактора. При раздражении нейрорецепторов возникает рефлекторный потенциал, энергия раздражения. Возбуждение этих рецепторов является пусковым фактором в развитии болезни. Нейрорецепторы образуют рецепторные поля, рефлексогенные зоны. Эти зоны обладают специфичностью, избирательной чувствительностью. Например, рецепторы тонкого кишечника реагируют на брюшнотифозную палочку, рецепторы толстого кишечника — на дизентерийную палочку. Таким образом, специфичность рецепторов, рефлексогенных зон определяет локализацию болезненного процесса. Рецепторы и рефлексогенные зоны обладают также свойством реактивности. Реактивность рецепторов и рефлексогенных зон определяет качество патологии или болезни. Их раздражение ведет к развитию патологического рефлекса (например, возбуждение α-адренорецепторов норадреналином вызывает спазм коронарных сосудов, нарушение коронарного кровообращения и развитие ишемической болезни сердца).
2. Начало болезни может быть связано с повреждением афферентных (чувствительных) проводников. Раздражение этого звена отражает нарушение в тканях, органах и вызывает дистрофические изменения в них.
3. Центральное звено представлено ЦНС. Многие эмоциональные факторы влияют непосредственно на центральные структуры, вызывая нарушение функции и развитие болезни (например, неврозы, сердечно-сосудистые заболевания). Следствием действия этих стрессорных факторов является формирование в ЦНС патологической системы саморегуляции: развивается дисрегуляция, возникает патологическая доминанта, нарушается интегративная деятельность мозга. Эти процессы отражают психосоматические отношения, что послужило толчком для развития психосоматической медицины. Психосоматические нарушения определяют развитие большинства заболеваний.
4. Эфферентное звено — это регуляторное звено. Оно представлено симпатической и парасимпатической нервной системой. Происходит дисбаланс между этими системами. Преобладание симпатической нервной системы ведет к развитию сердечно-сосудистых заболеваний, преобладание парасимпатической нервной системы — к развитию аллергии, патологии желудочно-кишечного тракта.
Гуморальные механизмы.
Эти механизмы характеризуют участие гормонов, нейромедиаторов, биологически активных веществ. Гуморальные факторы взаимодействуют с клеточными рецепторами, оказывая на них прямое действие. Гуморальные механизмы определяют специфичность заболевания. Например, главным звеном в развитии сахарного диабета внепанкреатического типа является нарушение функции инсулиновых рецепторов. В развитии аллергических заболеваний большое место занимают биологически активные вещества.
Метаболические механизмы
Эти механизмы являются универсальными. Они могут быть первичным звеном в развитии болезни. В реализации этого механизма большую роль играют нарушение выработки энергии, накопление токсических веществ (свободных радикалов, аммиака, гидроперекисей липидов), нарушение биосинтетических процессов.
Патоиммунные механизмы.
Эти механизмы играют основную роль в аллергических реакциях, при иммунодефицитных состояниях. Но они играют роль и при других заболеваниях, являются вторичными. Например, при атеросклерозе. Реализация этого механизма осуществляется благодаря участию цитотоксических лимфоцитов и образованию патоиммунного комплекса «Антиген-Антитело».
Генетические механизмы.
Эти механизмы являются ведущими в развитии наследственных и хромосомных заболеваний. Но они также принимают участие в развитии других болезней, например, лейкозов, опухолей. Реализация этих механизмов осуществляется через конформацию системы «ДНК-РНК-белок».
Местные реакции повреждения(локализация)— ограничение болезненных явлений, связанное со специфичностью рефлексогенных зон, их избирательностью и реактивностью (фурункул).
Общие реакции повреждения(генерализация)-распространение болезненных явлений, связанное с нарушением общей реактивности ( сепсис, воспаление).
«Порочный круг»— замкнутый цикл пат. процессов, образующийся по принципу причинно-следственных отношений.
Пример «порочного круга» при кровопотере:
Кровопотеря( уменьшение ОЦК)
Уменьшение общей кислородной емкости крови
Острая сердечная недостаточность
3.Гипероксия: её роль в патологии. Гипербарическая оксигенация и её использование в медицине. Механизмы лечебного действия гипероксии, адаптационно-метаболическая теория (А.Н.Леонов).
источник
 Анемия Под определение «гемолитическая анемия» подпадает любая анемия, характеризующаяся преобладанием гемолиза (процессом распада эритроцитов) над эритропоэзом (процессом образования эритроцитов). Гемолиз может быть иммунный и неиммунный. Гемолитическая анемия бывает наследственные и приобретенные.
Анемия Под определение «гемолитическая анемия» подпадает любая анемия, характеризующаяся преобладанием гемолиза (процессом распада эритроцитов) над эритропоэзом (процессом образования эритроцитов). Гемолиз может быть иммунный и неиммунный. Гемолитическая анемия бывает наследственные и приобретенные.
- Наследственная форма гемолитической анемии, обусловленная нарушением мембраны эритроцитов
- Наследственная форма гемолитической анемии, обусловленная нарушением активности ферментов эритроцитов
- Наследственная форма гемолитической анемии, обусловленная нарушением синтеза или структуры гемоглобина
- Анемия, обусловленная влиянием антител
- Анемия, обусловленная изменением структуры мембраны, вызванной соматической мутацией
- Анемия, обусловленная механическим повреждением оболочки эритроцитов
- Анемия, вызванная химическим повреждением эритроцитов
- Анемия, вызванная дефицитом витаминов (фолиевой кислоты и цианокобаламина)
- Анемия, вызванная разрушением эритроцитов паразитами
Болезнь Минковского-Шоффара (наследственный микросфероцитоз) – группа наследственных гемолитических анемий, характеризующихся образованием микросфероцитов (шаровидных эритроцитов) и обусловленных дефектом протеинов цитоскелета эритроцитов. При этом эритроциты теряют часть мембраны, уменьшается соотношение площади поверхности к объему, в результате чего эритроцит превращается в микросфероцит. Как правило, патология наследуется по аутосомно-доминантному признаку. Распространенность наследственного микросфероцитоза составляет примерно 1 случай на 1000-4500 человек.
При наследственном микросфероцитозе генетические нарушения влияют на протеины цитоскелета, преимущественно на те, которые объединяют цитоскелет с мембраной эритроцита. У большинства больных отмечается значительный дефицит спектрина, и только в некоторых случаях этот дефицит обусловлен генетическими дефектами самого спектрина.
Главные признаки наследственного микросфероцитоза – анемия, желтуха, спленомегалия (увеличенная селезенка). Анемия возникает из-за внутриклеточного распада эритроцитов. Желтуха развивается посредством непрямой гипербилирубинемии, может быть непостоянной и, как правило, слабо выражена у детей раннего возраста. Повышенное содержание билирубина в желчи часто является причиной образования пигментных желчных камней (даже у детей). Увеличение селезенки (спленомегалия) отмечается практически во всех случаях. При системных инфекционных патологиях интенсивность гемолиза может увеличиваться, в результате чего развивается спленомегалия.
Тяжелые формы наследственного микросфероцитоза характеризуются деформацией скелета: изменение расположения зубов, акрокефалия (башенный череп), высокое верхнее небо, микрофтальмия (уменьшение глазного яблока). В некоторых случаях отмечаются укороченные мизинцы. Могут образовываться трофические язвы на ногах.
Наследственный микросфероцитоз сопровождается апластическими кризами, которые провоцируются инфекцией (особенно парвовирусной).
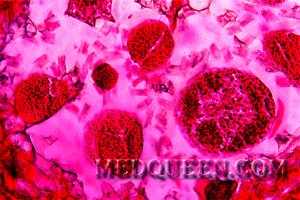
Микросфероцитоз – характерное изменение формы эритроцитов при этой патологии. При анализе мазка крови в биологическом материале наблюдаются микросфероциты в виде мелких клеток без центрального просветления (см рисунок 1). Отметим, что обнаружение микросфероцитов в мазках не всегда является признаком наследственного сфероцитоза.
Рисунок 1. Наследственный микросфероцитоз. Микросфероциты в мазке периферической крови (окр. по Романовскому-Гимзе, ув. ×100)
Такой признак обнаруживается при аутоиммунной гемолитической анемии с неполными тепловыми агглютинами, при наследственных дизэритропоэтической анемии. Средний объем эритроцитов, как правило, остается в норме или незначительно снижен. Показатель среднего содержания гемоглобина в эритроцитах в норме или незначительно повышен. Средняя концентрация гемоглобина в эритроцитах повышена почти у 50% пациентов.
Количественным показателем сферичности эритроцитов является осмотическая устойчивость (она снижена). Уровень ретикулоцитов в крови при гемолитическом кризе может значительно повышаться. Миелограмма показывает резкое раздражение красного ростка. Дифференциальный диагноз проводят с аутоиммунной гемолитической анемией, для которой характерна положительная проба Кумбса, отсутствие этой патологии среди родственников пациента и отсутствие данных о начале заболевания в детском возрасте.
Основной метод лечения анемии при наследственном микросфероцитозе – спленэктомия, с помощью которой устраняется анемия; при этом нельзя устранить морфологический дефект эритроцитов.
Наследственная гемолитическая анемия, обусловленная дефицитом глюкозо-6-фосфат дегидрогеназы эритроцитов – наиболее распространенная ферментопатия эритроцитов из группы ферментопатий пентозофосфатного пути метаболизма глюкозы. Глюкозо-6-фосфатдегидрогеназа эритроцитов – олигомер (в зависимости от условий может быть димер или тетрамер), который состоит из субъединиц с молекулярной массой 56 000 D. По данным ВОЗ (Всемирной организации здравоохранения) во всем мире количество людей, страдающих этой патологией, составляет более 200 млн. Наиболее широкое распространение этого заболевания характерно для Средиземноморского региона (Сицилия, Греция, Сардиния), негроидной расы, жителей Ближнего и Дальнего востока.
Клиническая картина при наследственной форме гемолитической анемии полиморфна: степень тяжести патологии может колебаться от гемолитической анемии, возникающей спонтанно после рождения, до гемолитических кризов. Гемолитический криз, который может провоцироваться метаболическим ацидозом или гипогликемией, развивается за несколько часов. В тяжелых случаях у больного развивается гемоглобинурия и шок. Также наблюдаются желтуха, моча приобретает бурый или черный цвет, одышка, диарея, рвота, снижение артериального давления, развивается тяжелая анемия, увеличиваются печень (гепатомегалия) и селезенка (спленомегалия).
Тяжелый гемолитический криз может спровоцировать развитие ДВС-синдрома (диссеминированного внутрисосудистого свертывания крови). Некоторые пациенты не переносят конские бобы (Viciafaba), после употребления которых происходит молниеносное развитие гемолитического криза (это явление также известно, как фовизм или примахиновая анемия).
Дефицит глюкозо-6-фосфат дегидрогеназы эритроцитов необходимо подозревать во всех случаях острого гемолиза, особенно у лиц негроидной расы и жителей средиземноморского региона. Диагноз подтверждается путем проведения лабораторных анализов. Острый гемолиз характеризуется быстрым снижением гематокрита с одновременным повышением уровня гемоглобина и непрямого гемоглобина, а также снижением уровня гаптоглобина. Анализ мазка крови показывает наличие фрагментов эритроцитов. Основой диагностики считается качественное (при необходимости – количественное) определение активности глюкозо-6-фосфат дегидрогеназы эритроцитов. У пациентов с вариантом «А-» явление аномального гемолиза проходит, как правило, самостоятельно – такие больные не нуждаются в специальном лечении. В случае развития тяжелого гемолитического криза необходимо проводить форсированный диурез, профилактику ДВС-синдрома, плазмаферез (с целью удаления продуктов гемолиза).
В случае возникновения качественной гемоглобинопатии происходит изменение аминокислотной последовательности цепей глобина. Талассемия (количественная гемоглобинопатия) характеризуется снижением образования цепей глобина без изменения их цепей. Нужно отметить, что разница между качественной и количественной гемоглобинопатиями не абсолютна.
Талассемия (анемия Кули) – группа патологий, обусловленных генетическим нарушением синтеза одной из цепей глобина. В норме процесс синтеза глобиновых цепей сбалансирован, поэтому свободных цепей глобина нет. В случае нарушения синтеза одной из цепей глобина баланс нарушается, образуются лишние цепи, которые агрегируют и откладываются в эритрокариоцитах. Среди жителей Средиземноморья наиболее распространена β-талассемия.
«Большая талассемия» (болезнь Кули, β-талассемия) – наследственная гемолитическая анемия, впервые описанная американскими педиатрами-гематологами Томасом Бентоном Кули (Thomas Benton Cooley) и Ли (P. Lee) в статье «Серия случаев спленомегалии у детей с анемией и необычными изменениями костей» («A Series of Cases of Splenomegaly in Children, with Anemia and Peculiar Bone Changes»), где были приведены случаи у выходцев из стран Средиземноморья. Для анемии Кули характерна тяжелая степень течения с самого детства, задержка роста и изменения костей в результате увеличения объема костного мозга, возникающие в случае отсутствия соответствующего лечения). Также при этой патологии у больного наблюдаются гепатомегалия, спленомегалия, гиперспленизм, деформации черепа (монголоидное лицо, башенный череп); желтуха, бледность и отложение меланина придают коже особый медный оттенок. Кроме этого, наблюдается перегрузка железом сердца, легких, печени, поджелудочной железы и других органов эндокринной системы, переломы костей, сдавления периферических нервов, разного рода инфекционные осложнения.
Результаты лабораторных исследований периферической крови показывают гипохромную анемию, ретикулоцитоз, мишеневидные эритроциты (см рис 2-4).
Рисунок 02. Анемия Кули (большая талассемия). Периферическая кровь. Микроцитоз, выраженная гипохромия, мишеневидные нормобласты и эритроциты (окр. по Романовскому-Гимзе, ув. ×100)
Рисунок 03. Анемия Кули (большая талассемия). Периферическая кровь (окр. по Романовскому-Гимзе, ув. ×50)
Рисунок 04. Анемия Кули (большая талассемия). Периферическая кровь. Множественные мишеневидные эритроциты (окр. по Романовскому-Гимзе, ув. ×100)
Миелограмма демонстрирует раздражение «красного ростка» и повышение количества сидеробластов. Также наблюдается повышение осмотической резистентности эритроцитов и количества билирубина за счет непрямой фракции. В крови повышается содержание железа и ферритина, развивается гемосидероз (чрезмерное отложение гемосидерина в тканях) внутренних органов. При гомозиготной β-талассемии необходимо проводить пренатальную диагностику – забор клеток плода из амниотической жидкости на предмет выявления мутации генов, отвечающих за кодирование β-цепи глобина, с применением метода полимеразной цепной реакции.
Без соответствующего лечения больные анемией Кули умирают в детском возрасте. Продлить жизнь, предупредить деформации костей и задержку роста можно путем регулярных трансфузий эритроцитарной массы (лучше переливать отмытые или размороженные эритроциты) при условии поддержания достаточно высокого уровня гемоглобина. В случае значительной спленомегалии и явлениях гиперспленизма больному показана спленэктомия (удаление селезенки). С целью предотвращения развития гемосидероза пациентам периодически назначают Деферазирокс (Эксиджад) или Дефероксамин (Десферал). Излечение возможно при аллогенной трансплантации костного мозга.
Серповидноклеточная анемия обусловлена носительством гемоглобина, который меняет свою структуру в условиях гипоксии. Самой распространенной аномалией структуры гемоглобина является гемоглобинопатия Sα2β26 глу+вал. При гомозиготном носительстве можно говорить о серповидноклеточной анемии; при гетерозиготном носительстве – серповидноклеточная аномалия. Патология наследуется по аутосомно-доминантному признаку. При серповидноклеточной анемии наблюдается мутация, в результате которой в цепи глобина глутаминовая кислота заменяется валином. В результате растворимость гемоглобина S при отдаче кислорода снижается, что приводит к образованию геля.
Серповидноклеточная анемия наиболее распространена среди населения Центральной Африки, Турции, Индии, Кубы. У больных диагностируется анемия, тромботические осложнения, поражения костей и суставов (отмечаются некрозы плечевой и бедренной костей). Кроме этого, тромбозы осложняются инфарктами (сердца, легких, почек, селезенки, головного мозга), приступами сильной боли в области живота. У детей отмечаются нарушения физического (отставание в росте) и полового развития, ночное недержание мочи, нарушение зрения (тромбозы сосудов сетчатки). Также могут развиваться гемолитический, апластический и секвестрационные кризы, при этом в селезенке происходит резкое накопление эритроцитов, что вызывает гиповолемический шок и резкое снижение уровня гемоглобина.
Для анализов крови при апластической анемии характерны низкий уровень гемоглобина, наличие серповидных эритроцитов (рисунок 5), базофильная пунктация эритроцитов, их мишеневидность, повышение уровня ретикулоцитов и непрямого билирубина. Миелограмма демонстрирует раздражение «красного ростка».
Рисунок 5. Серповидноклеточная анемия. Периферическая кровь. Серповидные и мишеневидные эритроциты. выраженная гипохромия эритроцитов (окр. по Романовскому-Гимзе, ув. ×100)
В качестве лечения применяют адекватную инфузионную терапию, переливания эритроцитарной массы, оксигенотерапии.
К приобретенным гемолитическим анемиям относится группа заболеваний разного патогенеза, которые объединяет внутрисосудистый гемолиз (гемолиз эритроцитов в периферической крови). В зависимости от механизма эритролиза приобретенная гемолитическая анемия может носить иммунный и неиммунный характер. Но, несмотря на разные патогенетические механизмы, клинические признаки этих анемий часто совпадают.
Гемолитическая анемия у пациентов с протезированными клапанами сердца и сосудами развивается примерно в 10% случаев при протезированном аортальном клапане. При использовании стеллитовых запирательных элементов частота гемолиза незначительно увеличивается (по сравнению с селиконовыми). Также некоторое увеличение частоты гемолиза отмечается при наличии околоклапанной регургитации и при малом диаметре клапана. Биопротезы (свиные клапаны) в редких случаях являются причиной механического гемолиза. Гораздо реже причиной гемолиза может быть также протезированный митральный клапан, так как трансклапанный градиент давления в этом случае ниже.
Гемолиз протезированными клапанами происходит в результате одновременного действия сразу нескольких факторов:
- Значительная сила сдвига, которая при турбулентном токе крови действует на мембрану эритроцитов, особенно когда под высоким давлением кровь проходит через маленькое отверстие (например, при околоклапанной регургитации)
- Отложения фибрина на участках неплотного прилегания кольца клапана к тканям сердца
- Прямое механическое повреждение эритроцитов при закрытии запирательного элемента
Значительное разрушение эритроцитов может наблюдаться после закрытия дефекта межпредсердной перегородки типа ostium primum заплатой из синтетического материала. Умеренное сокращение жизни эритроцитов с легкой анемией или без нее может наблюдаться при значительном обызвествлении аортального клапана. Механический гемолиз обнаруживается также у пациентов, перенесших аортокоронарное и аортобедренное шунтирование.
Тяжелые случаи механического гемолиза сопровождаются тяжелой анемией, ретикулоцитозом, обнаруживаются фрагментированные эритроциты (шизоциты), гемоглобинемия и гемоглобинурия, повышается активность лактатдегидрогеназы, снижается уровень гаптоглобина. Выведение железа из организма с мочой в виде гемосидерина или гемоглобина может вызвать дефицит железа в организме. В случае развития дефицита железа пациенту назначается пероральный прием препаратов железа. Терапия препаратами железа способствует повышению уровня гемоглобина и способствует снижению сердечного выброса и снижению интенсивности гемолиза. Отметим, что ограничение физической активности также способствуют снижению интенсивности распада эритроцитов. Если предпринимаемые меры не приводят к желаемому результату, нужно полностью устранить околоклапанную регургитацию или заменить протез.
источник
Гемолитическая анемия – патология эритроцитов, отличительным признаком которой является ускоренное разрушение красных кровяных телец с высвобождением повышенного количества непрямого билирубина. Для данной группы заболеваний типично сочетание анемического синдрома, желтухи и увеличения размеров селезенки. В процессе диагностики исследуется общий анализ крови, уровень билирубина, анализ кала и мочи, УЗИ органов брюшной полости; проводится биопсия костного мозга, иммунологические исследования. В качестве методов лечения используется медикаментозная, гемотрансфузионная терапия; при гиперспленизме показана спленэктомия.
Гемолитическая анемия (ГА) — малокровие, обусловленное нарушением жизненного цикла эритроцитов, а именно преобладанием процессов их разрушения (эритроцитолиза) над образованием и созреванием (эритропоэзом). Данная группа анемий очень обширна. Их распространенность неодинакова в различных географических широтах и возрастных когортах; в среднем патология встречается у 1% населения. Среди прочих видов анемий на долю гемолитических приходится 11%. Патология характеризуется укорочением жизненного цикла эритроцитов и их распадом (гемолизом) раньше времени (через 14-21 день вместо 100-120 суток в норме). При этом разрушение эритроцитов может происходить непосредственно в сосудистом русле (внутрисосудистый гемолиз) или в селезенке, печени, костном мозге (внесосудистый гемолиз).
Этиопатогенетическую основу наследственных гемолитических синдромов составляют генетические дефекты мембран эритроцитов, их ферментных систем либо структуры гемоглобина. Данные предпосылки обусловливают морфофункциональную неполноценность эритроцитов и их повышенное разрушение. Гемолиз эритроцитов при приобретенных анемиях наступает под влиянием внутренних факторов или факторов окружающей среды, среди которых:
- Аутоиммунные процессы. Образование антител, агглютинирующих эритроциты, возможно при гемобластозах (остром лейкозе, хроническом лимфолейкозе, лимфогранулематозе), аутоиммунной патологии (СКВ, неспецифическом язвенном колите), инфекционных заболеваниях (инфекционном мононуклеозе, токсоплазмозе, сифилисе, вирусной пневмонии). Развитию иммунных гемолитических анемий могут способствовать посттрансфузионные реакции, профилактическая вакцинация, гемолитическая болезнь плода.
- Токсическое действие на эритроциты. В ряде случаев острому внутрисосудистому гемолизу предшествует отравление мышьяковистыми соединениями, тяжелыми металлами, уксусной кислотой, грибными ядами, алкоголем и др. Вызывать разрушение клеток крови может прием определенных лекарств (противомалярийных препаратов, сульфаниламидов, производных нитрофуранового ряда, анальгетиков).
- Механическое повреждение эритроцитов. Гемолиз эритроцитов может наблюдаться при тяжелых физических нагрузках (длительной ходьбе, беге, лыжном переходе), при ДВС-синдроме, малярии, злокачественной артериальной гипертензии, протезировании клапанов сердца и сосудов, проведении гипербарической оксигенации, сепсисе, обширных ожогах. В этих случаях под действием тех или иных факторов происходит травматизация и разрыв мембран изначально полноценных эритроцитов.
Центральным звеном патогенеза ГА является повышенное разрушение эритроцитов в органах ретикулоэндотелиальной системы (селезенке, печени, костном мозге, лимфатических узлах) или непосредственно в сосудистом русле. При аутоиммунном механизме анемии происходит образование антиэритроцитарных АТ (тепловых, холодовых), которые вызывают ферментативный лизис мембраны эритроцитов. Токсические вещества, являясь сильнейшими окислителями, разрушают эритроцит за счет развития метаболических, функциональных и морфологических изменений оболочки и стромы красных кровяных телец. Механические факторы оказывают прямое воздействие на клеточную мембрану. Под влиянием этих механизмов из эритроцитов выходят ионы калия и фосфора, а внутрь поступают ионы натрия. Клетка разбухает, при критическом увеличении ее объема наступает гемолиз. Распад эритроцитов сопровождаются развитием анемического и желтушного синдромов (так называемой «бледной желтухой»). Возможно интенсивное окрашивание кала и мочи, увеличение селезенки и печени.
В гематологии гемолитические анемии подразделяются на две большие группы: врожденные (наследственные) и приобретенные. Наследственные ГА включают следующие формы:
- эритроцитарные мембранопатии (микросфероцитоз – болезнь Минковского-Шоффара, овалоцитоз, акантоцитоз) – анемии, обусловлены структурными аномалиями мембран эритроцитов
- ферментопении (энзимопении) – анемии, вызванные дефицитом тех или иных ферментов (глюкозо-6-фосфатдегидрогеназы, пируваткиназы и др.)
- гемоглобинопатии— анемии, связанные с качественными нарушениями структуры гемоглобина или изменением соотношения его нормальных форм (талассемия, серповидно-клеточная анемия).
Приобретенные ГА подразделяются на:
- мембранопатии приобретенные (пароксизмальная ночная гемоглобинурия – б-нь Маркиафавы-Микели, шпороклеточная анемия)
- иммунные (ауто- и изоиммунные) – обусловлены воздействием антител
- токсические – анемии, обусловленные воздействием химических веществ, биологических ядов, бактериальных токсинов
- механические — анемии, вызванные механическим повреждением структуры эритроцитов (тромбоцитопеническая пурпура, маршевая гемоглобинурия)
Наиболее распространенной формой данной группы анемий является микросфероцитоз, или болезнь Минковского-Шоффара. Наследуется по аутосомно-доминантному типу; обычно прослеживается у нескольких представителей семьи. Дефектность эритроцитов обусловлена дефицитом в мембране актомиозиноподобного белка и липидов, что приводит к изменению формы и диаметра эритроцитов, их массивному и преждевременному гемолизу в селезенке. Манифестация микросфероцитарной ГА возможна в любом возрасте (в младенчестве, юношестве, старости), однако обычно проявления возникают у детей старшего возраста и подростков. Тяжесть заболевания варьирует от субклинического течения до тяжелых форм, характеризующихся часто повторяющимися гемолитическими кризами. В момент криза нарастает температура тела, головокружение, слабость; возникают боли в животе и рвота.
Основным признаком микросфероцитарной гемолитической анемии служит желтуха различной степени интенсивности. Вследствие высокого содержания стеркобилина кал становится интенсивно окрашенным в темно-коричневый цвет. У пациентов с болезнь Минковского-Шоффара наблюдается склонность к образованию камней в желчном пузыре, поэтому часто развиваются признаки обострения калькулезного холецистита, возникают приступы желчной колики, а при закупорке холедоха конкрементом — обтурационная желтуха. При микросфероцитозе во всех случаях увеличена селезенка, а у половины пациентов – еще и печень. Кроме наследственной микросфероцитарной анемии, у детей часто встречаются другие врожденные дисплазии: башенный череп, косоглазие, седловидная деформация носа, аномалии прикуса, готическое нёбо, полидактилия или брадидактилия и пр. Пациенты среднего и пожилого возраста страдают трофическими язвами голени, которые возникают в результате гемолиза эритроцитов в капиллярах конечностей и плохо поддаются лечению.
Энзимопенические анемии связаны с недостатком определенных ферментов эритроцитов (чаще — Г-6-ФД, глутатион-зависимых ферментов, пируваткиназы и др). Гемолитическая анемия может впервые заявлять о себе после перенесенного интеркуррентного заболевания или приема медикаментов (салицилатов, сульфаниламидов, нитрофуранов). Обычно заболевание имеет ровное течение; типична «бледная желтуха», умеренная гепатоспленомегалия, сердечные шумы. В тяжелых случаях развивается ярко выраженная картина гемолитического криза (слабость, рвота, одышка, сердцебиение, коллаптоидное состояние). В связи с внутрисосудистым гемолизом эритроцитов и выделением гемосидерина с мочой последняя приобретает темный (иногда черный) цвет. Особенностям клинического течения гемоглобинопатий — талассемии и серповидно-клеточной анемии посвящены самостоятельные обзоры.
Среди различных приобретенных вариантов чаще других встречаются аутоиммунные анемии. Для них общим пусковым фактором выступает образование антител к антигенам собственных эритроцитов. Гемолиз эритроцитов может носить как внутрисосудистый, так и внутриклеточный характер. Гемолитический криз при аутоиммунной анемии развивается остро и внезапно. Он протекает с лихорадкой, резкой слабостью, головокружением, сердцебиением, одышкой, болями в эпигастрии и пояснице. Иногда острым проявлениям предшествуют предвестники в виде субфебрилитета и артралгий. В период криза стремительно нарастает желтуха, не сопровождающаяся кожным зудом, увеличивается печень и селезенка. При некоторых формах аутоиммунных анемий больные плохо переносят холод; в условиях низких температур у них может развиваться синдром Рейно, крапивница, гемоглобинурия. Вследствие недостаточности кровообращения в мелких сосудах возможны осложнения в виде гангрены пальцев ног и рук.
Токсические анемии протекают с прогрессирующей слабостью, болями в правом подреберье и поясничной области, рвотой, гемоглобинурией, высокой температурой тела. Со 2-3 суток присоединяется желтуха и билирубинемия; на 3-5 сутки возникает печеночная и почечная недостаточность, признаками которых служат гепатомегалия, ферментемия, азотемия, анурия. Отдельные виды приобретенных гемолитических анемий рассмотрены в соответствующих статьях: «Гемоглобинурия» и «Тромбоцитопеническая пурпура», «Гемолитическая болезнь плода».
Каждый вид ГА имеет свои специфические осложнения: например, ЖКБ – при микросфероцитозе, печеночная недостаточность – при токсических формах и т.д. К числу общих осложнений относятся гемолитические кризы, которые могут провоцироваться инфекциями, стрессами, родами у женщин. При остром массивном гемолизе возможно развитие гемолитической комы, характеризующейся коллапсом, спутанным сознанием, олигурией, усилением желтухи. Угрозу жизни больного несут ДВС-синдром, инфаркт селезенки или спонтанный разрыв органа. Неотложной медицинской помощи требуют острая сердечно-сосудистая и почечная недостаточность.
Определение формы ГА на основе анализа причин, симптоматики и объективных данных относится к компетенции гематолога. При первичной беседе выясняется семейный анамнез, частота и тяжесть протекания гемолитических кризов. В процессе осмотра оценивается окраска кожных покровов, склер и видимых слизистых, производится пальпация живота для оценки величины печени и селезенки. Сплено- и гепатомегалия подтверждается при проведении УЗИ печени и селезенки. Лабораторный диагностический комплекс включает:
- Исследование крови. Изменения в гемограмме характеризуются нормо- или гипохромной анемией, лейкопенией, тромбоцитопенией, ретикулоцитозом, ускорением СОЭ. В биохимических пробах крови определяется гипербилирубинемия (увеличение фракции непрямого билирубина), увеличение активности лактатдегидрогеназы. При аутоиммунных анемиях большое диагностическое значение имеет положительная проба Кумбса.
- Анализы мочи и кала. Исследование мочи выявляет протеинурию, уробилинурию, гемосидеринурию, гемоглобинурию. В копрограмме повышено содержание стеркобилина.
- Миелограмму. Для цитологического подтверждения выполняется стернальная пункция. Исследование пунктата костного мозга обнаруживает гиперплазию эритроидного ростка.
В процессе дифференциальной диагностики исключаются гепатиты, цирроз печени, портальная гипертензия, гепатолиенальный синдром, порфирии, гемобластозы. Пациента консультируют гастроэнтеролог, клинический фармаколог, инфекционист и другие специалисты.
Различные формы ГА имеют свои особенности и подходы к лечению. При всех вариантах приобретенной гемолитической анемии необходимо позаботиться об устранении влияния гемолизирующих факторов. Во время гемолитических кризов больным необходимы инфузии растворов, плазмы крови; витаминотерапия, по необходимости – гормоно- и антибиотикотерапия. При микросфероцитозе единственно эффективным методом, приводящим к 100 % прекращению гемолиза, является спленэктомия.
При аутоиммунной анемии показана терапия глюкокортикоидными гормонами (преднизолоном), сокращающая или прекращающая гемолиз. В некоторых случаях требуемый эффект достигается назначением иммунодепрессантов (азатиоприна, 6-меркаптопурина, хлорамбуцила), противомалярийных препаратов (хлорохина). При резистентных к медикаментозной терапии формах аутоиммунной анемии выполняется спленэктомия. Лечение гемоглобинурии предполагает переливание отмытых эритроцитов, плазмозаменителей, назначение антикоагулянтов и антиагрегантов. Развитие токсической гемолитической анемии диктует необходимость проведения интенсивной терапии: дезинтоксикации, форсированного диуреза, гемодиализа, по показаниям – введение антидотов.
Течение и исход зависят от вида анемии, тяжести протекания кризов, полноты патогенетической терапии. При многих приобретенных вариантах устранение причин и полноценное лечение приводит к полному выздоровлению. Излечения врожденных анемий добиться нельзя, однако возможно достижение длительной ремиссии. При развитии почечной недостаточности и других фатальных осложнений прогноз неблагоприятен. Предупредить развитие ГА позволяет профилактика острых инфекционных заболеваний, интоксикаций, отравлений. Запрещается бесконтрольное самостоятельное использование лекарственных препаратов. Необходимо тщательная подготовка пациентов к гемотрансфузиям, вакцинации с проведением всего комплекса необходимых обследований.
источник
Гемолитическая анемия – это заболевание, которое состоит из нескольких патологий, общей симптоматикой которых является усиленное разрушение эритроцитов в сосудистом русле. Данный вид болезни занимает более 5% всей анемии. Учёные продолжают изучать гемолитические анемии, классификация которых делит формы заболевания на наследственную и приобретённую. В статье будет рассмотрена тема, почему происходит развитие этого недуга.
Наследственная гемолитическая анемия развивается на фоне врождённых дефектов, которые затрагивают гемоглобин, ферменты, мембраны эритроцитов. Данный вид также делят на подвиды:
При гемоглобинопатии происходит генетический дефект, что приводит к нарушению строения гемоглобина. Во время этого процесса осуществляется образование аномального гемоглобина S, что, в свою очередь, изменяет эритроциты, которые становятся удлинёнными по форме. Болезнь приводит к затруднению эритроцитного прохождения по мелким сосудам, через определённое время из-за этого происходит их закупорка и начинается развитие многочисленных инфарктов.
Данная наследственная гемолитическая анемия развивается у детей. Проявляется недуг такой симптоматикой:
- бледнее кожа;
- появляется быстрая утомляемость;
- задержка роста;
- повышается чувствительность к различным инфекциям (сальмонеллёз и др.);
- трофические язвы голеней;
- ухудшение реологических особенностей крови;
- кровоизлияние в сетчатку.
Гемолитическая анемия у детей, протекающая в хронической форме, приводит к желтухе, желчнокаменной болезни, остеомелиту, асептическому некрозу головки бедренной кости. К тяжёлым осложнениям относят инсульт, болевой криз, во время которого у человека появляются болевые ощущения в области спины, рёбер, конечностей. Такие боли продолжаются на протяжении нескольких дней или недель. К ним присоединяется лихорадка, при этом наблюдается нормальная концентрация гемоглобина. Также возможным осложнением является острый синдром грудной клетки.
Иногда гемолитическая анемия у новорождённых и у людей разного возраста, может вызывать секвестрационные кризы. Этот процесс характеризуется кровяным депонированием в селезёнке, на фоне чего возникают артериальная гипотония, шок, падает уровень гемоглобина.
В редких случаях отмечается гемолитический криз, а если имеется вирусная инфекция, может развиться апластический криз, который приводит к резкому снижению гемоглобина, уменьшению ретикулоцитов.
 Для усиления эффекта Гидроксимочевины добавляют прием Эритропоэтина
Для усиления эффекта Гидроксимочевины добавляют прием Эритропоэтина
При постановке диагноза гемолитическая анемия, лечение должно быть своевременным. Обычно при терапии данного вида заболевания прибегают к «Гидроксимочевине». Благодаря этому препарату осуществляется повышение фетального гемоглобина, а также снижение гемолиза. Чтобы усилить эффективность этого средства, врачи назначают больному приём «Эритропоэтина». Важно отметить, что данная методика применима лишь во время лечения тяжёлой формы недуга.
Основной задачей терапии считается предотвращение острых и хронических осложнений. Грудничкам и детям до 5 лет вводят менингококковую, «Haemophilus influenza» типа В вакцину. В качестве профилактической меры детям старшего возраста необходим приём «Пенициллина» 125 – 250 мг. каждые сутки, а при проявлении лихорадки следует провести интенсивные противомикробные процедуры. Больным, у которых наблюдается хроническое течение болезни, нужно употреблять ежедневно фолиевую кислоту по 1 мг.
Болевой криз принуждает к проведению обезболивающей терапии с помощью анальгетиков. Считается, что самым эффективным является длительная инфузия «Морфина». Врачи учитывают особенность данного препарата и склонность пациентов к рецидивам болевого синдрома, чтобы исключить наркотическую зависимость.
Во время острого синдрома грудной клетки осуществляются кислородная ингаляция, инфузионные процедуры, приём антибиотиков («Эритромицин», «Цефтриаксон»). Если анализ крови показывает менее 60 мм. рт. ст. показателя РаО2, врачи принимают решение провести обменное переливание крови.
Перед лечением остеомелита пациенту проводят биопсию. Если данные процедуры не дали положительного результата, используют трансфузионную терапию, а также пересадку полнослойных кожных лоскутов.
Шанс на излечение от этой разновидности гемологической анемии (код по МКБ10 – международная классификация болезней – D55 – D59) даёт аллотрансплатация костного мозга. Но на сегодняшний день методика не имеет широкого применения.
Данный вид заболевания характеризуется дефицитом активности эритроцитарных ферментов. Ген, провоцирующий болезнь, наследуется по мужской линии, чаще всего у африканцев, китайцев и жителей Средиземноморья. У женского пола недуг встречается нечасто.
Гемолитическая анемия, симптомы которой развиваются быстро, проявляется гемолитическим кризом. Учёные отмечают, что симптоматика недуга может наблюдаться уже через несколько часов после воздействия провоцирующего фактора:
- инфекционные болезни;
- бобовая пища;
- медицинские препараты (« Аспирин», «Хинин», «Сульфаниламид» и др.).
 Аспирин может спровоцировать гемолитическую анемию
Аспирин может спровоцировать гемолитическую анемию
При терапии заболевания врачи ведут борьбу с инфекциями, исключают приём некоторых лекарств, запрещают бобы. Проводится гемотрансфузия, но при строгих показателях.
Такое заболевание характеризуется структурной аномалией белков эритроцитов, на фоне чего происходит нарушение их мембран, развивается внесосудистый гемолиз. Данная разновидность анемии также делится на формы:
- Микросфероцитарная гемолитическая анемия передаётся в большинстве случаев аутосомно доминантным типом, во время которого наблюдается дефект цитоскелета. В результате недуга эритроциты превращаются в микросфероциты, диаметр которых менее 7 мкм. Болезнь проявляется в любом возрасте, даже у новорождённых. К основной симптоматике относят желтуху, проявляющуюся наличием пигментных камней в желчном пузыре, спленомегалией, гиперплазией. Анализ крови обнаруживает микросфероциты. Спонтанное ухудшение состояния вызвано гемолизом, происходящим в течение двух суток. Этот процесс можно остановить с помощью введения в кровь глюкозы. Выздоравливает пациент после спленэктомии, а чтобы микросфероцитарная гемолитическая анемия не повторилась, ему назначают приём фолиевой кислоты на протяжении длительного периода времени.
- Овалоцитоз наследуется, как и при состоянии микросфероцитоза, обычно его обнаруживают случайно во время исследования периферической крови. При недуге может наблюдаться сниженная концентрация гемоглобина. Терапия этого вида подобна методам, как при микросфероцитозе.
- Стомацитоз меняет эритроциты, они становятся выпуклыми с одной стороны и вогнутыми с другой, также у них образуется щелевидное пространство. Эритроциты могут быть гипергидратированными и дегидратированными. Клиническая картина недуга может характеризоваться лёгким гемолизом.
Гемолитическая анемия приобретённого типа, причины которой могут быть разными, также делится на виды:
- иммунная гемолитическая анемия;
- механическая;
- микроангиопатическая.
Аутоиммунная гемолитическая анемия у взрослого человека развивается из-за выработки аутоантител к антигенам эритроцитов. Острое развитие недуга проявляется одышкой, общей слабостью, ощущается сердцебиение, появляются болевые ощущения в области сердца, поясницы, повышается температура, может проявиться желтуха. Если иммунная гемолитическая анемия имеет хроническое течение, к этому состоянию добавляется увеличение селезёнки или печени. Для диагностики аутоиммунных анемий прибегают к пробе Кумбса. В свою очередь, иммунная гемолитическая анемия делится на несколько типов:
- Аутоиммунная гемолитическая анемия с антителами тепловыми чаще диагностируется у женского пола. Это заболевание подразделяют на лекарственное и идиопатическое, оно является осложнением гемобластозов. Помимо проявления основными признаками, при недуге наблюдаются спленомегалия, обмороки, болевые ощущения в области грудной клетки.
Аутоиммунная гемолитическая анемия в лёгкой форме не нуждается в терапии, при средней тяжести и тяжёлой лечение заключается на устранение причины развития данного заболевания. Так, в течение нескольких недель необходимо употреблять глюкокортикоиды, через определённое время дозировка лекарства должна быть снижена, затем на протяжении еще нескольких месяцев следует продолжать приём, при этом постепенно снижая дозировку. Такая терапия помогает поправить здоровье 80% пациентов, которым поставили диагноз иммунная гемолитическая анемия, но у половины из них через некоторое время происходит рецидив.
- Аутоиммунная гемолитическая анемия с антителами холодовыми имеет тесную связь с инфекциями, также лимфопролиферативным состоянием. К главному симптому недуга относят повышенную чувствительность к холоду. Так, при заболевании синеют и белеют пальцы конечностей, кончик носа, ушей. Иммунная гемолитическая анемия характеризуется расстройством периферического кровообращения.
- К болезни относят и пароксизмальную холодную гемоглобинурию. Такой вид считается редким и развивается в результате перенесённого вируса. Симптоматика даёт о себе знать лишь после пребывания на морозе. Приступ проявляется ознобом, лихорадкой, болью в спине, нижних конечностях, животе, голове, а также слабостью, гемоглобинурией. Главной задачей терапии считается предупреждение переохлаждения. Если недуг протекает в хронической форме, доктор назначает «Преднизолон», иммунодепрессанты.
 К аутоимунной гемолитической анемии относится пароксизмальная ночная гемоглобинурия, для профилактики назначают Преднизолон
К аутоимунной гемолитической анемии относится пароксизмальная ночная гемоглобинурия, для профилактики назначают Преднизолон
- Лекарственный тип развивается на фоне приёма медицинских средств, которые делят на группы. В первую входят лекарства, провоцирующие недуг, симптоматика схожа с аутоиммунным заболеванием типа с тепловыми антителами. Большинство пациентов страдают от данной болезни, образующейся в результате приёма «Метилдофа». Во вторую группу входят средства, абсорбирующиеся на эритроцитной поверхности – «Пенициллин» и другие антибиотики, схожие по структуре. К третьей группе относят препараты, которые провоцируют образование специфических антител IgM.
Учёные рассматривают причины, по которым возникает малокровие данного типа:
- Во время эритроцитного движения по мелким сосудам, которые расположены над костными выступами, эритроциты подвергаются сдавливанию, другими словами, происходит маршевая гемоглобинурия.
- В процессе преодоления градиента давления на сердечных, сосудистых протезированных клапанах.
- Выполняя движение по мелким сосудам, стенки которых отличаются возможностью изменяться.
Данный тип даёт о себе знать после ходьбы, бега на протяжении длительного времени, выполнения тренировки по тяжёлой атлетике и каратэ. По медицинской статистике, около 10% пациентов с протезированными клапанами страдают от болезни. Лечить механический недуг просто. Для этого снижают дефицит железа, ограничивают физическую нагрузку.
Разновидность болезни возникает на фоне патологии стенок сосудов, гемолитико-уремического синдрома, а также при диссеминированном внутрисосудистом свёртывании. При терапии больному недугом следует прибегнуть к гемодиализу, глюкокортикоидам, плазмаферезу, свежезамороженной плазме.
Врачи постоянно напоминают, что легче заболевание предупредить, нежели лечить. Поэтому ведение здорового образа жизни поможет предотвратить развитие серьёзных болезней.
источник