Med-books.by — Библиотека медицинской литературы . Книги, аудиокниги по медицине. Банк рефератов. Медицинские рефераты. Всё для студента-медика .
Скачать бесплатно без регистрации или купить электронные и печатные бумажные медицинские книги (DJVU, PDF, DOC, CHM, FB2, TXT), истории болезней, рефераты, монографии, лекции, презентации по медицине.
Med-books.by — Библиотека медицинской литературы » Истории: Внутренние болезни (Терапия) » История болезни: Аутоиммунная гемолитическая анемия, ср. степени тяжести, протекающая с мегалобластоидностью костного мозга.
История болезни: Аутоиммунная гемолитическая анемия, ср. степени тяжести, протекающая с мегалобластоидностью костного мозга.
Ф.И.О.: _____________
Возраст: 02.10.1953
Пол: женский
Национальность: белоруска
Семейное положение: замужем
Место работы: пенсионерка
Домашний адрес: _________________
Дата поступления в клинику: 14.11.2013
Группа крови: А(II) Rh-
Диагноз направившей организации: Анемия
Диагноз при поступлении: Аутоиммунная гемолитическая анемия
Клинический диагноз: Аутоиммунная гемолитическая анемия, ср. степени тяжести, протекающая с мегалобластоидностью костного мозга. Кардиомиопатия смешанного генеза Н 1. АГ 2ст риск 4.
На момент поступления: общая слабость, головокружение, одышка при физических нагрузках и в покое, повышенную утомляемость.
На момент курации: общая слабость, одышка при физических нагрузках, утомляемость.
Заболела в августе 2013 года, когда почувствовала резкую слабость. Обратились к терапевту по месту жительства. Была госпитализирована в стационар(по направлению поликлиники № 4) в гематологическое отделение УЗ ВОКБ. Отмечалось улучшение после лечения и была выписана. Однако 20 сентября вернулась в стационар. Месяц проходила лечение и была выписана. 14 ноября вновь поступила в стационар. Причиной был низкий гемоглобин в ОАК, а также жалобы пациентки на общую слабость и усталость.
Паротит, менингит, АГ. Миома матки.
Родилась в срок. Росла и развивалась нормально. Замужем. Двое детей.
Из перенесенных заболеваний отмечает простудные заболевания, паротит, менингит, АГ, миома матки. Туберкулез, вирусный гепатит, онкологические и венерические заболевания отрицает.
Было проведено переливание крови (14.11.2013, 16.11.2013). Аллерго-анамнез не отягощен.
Жилищно-бытовые условия удовлетворительные, питание регулярное.
Не курит, алкоголь не употребляет.
Наследственный анамнез не отягощён.
Общее состояние удовлетворительное. Сознание ясное. Настроение спо-койное. Телосложение правильное. Рост 163 см., вес 72 кг. Кожа сухая, чистая, окраска кожных покровов бледная, эластичность кожи сохранена. Видимые слизистые бледные. Сыпи, расчёсов, петехий, рубцов нет.
Периферические лимфатические узлы (подчелюстные, околоушные, шей-ные, затылочные, над- и подключичные, подмышечные, паховые) не увеличены. Шея правильной формы, видимой пульсации сосудов нет. Щитовидная железа не увеличена, доли не пальпируются. Молочная железа правильной формы, при пальпации патологических образований не выявлено. Позвоночник обычной конфигурации, без патологических искривлений, движения- в полном объеме. Походка нормальная.
Костно-мышечно-суставная система
Мышечная система развита умеренно, тонус и сила мышц несколько снижены. Мышцы при пальпации безболезненны. Ограничения движения в суставах нет.
Система органов дыхания
Грудная клетка нормостенической формы. Дыхание через нос. Число дыхательных движений 18 в минуту. При сравнительной перкуссии звук над обоими легкими ясный легочный. Тип дыхания — грудной. Форма грудной клетки правильная. При аускультации дыхание везикулярное. Границы легких в пределах нормы. Дыхание через нос, свободное. Тип дыхания грудной. Число дыханий – 16 в мин. Дыхание ритмичное.
Система органов кровообращения
При осмотре верхушечный толчок не виден. Патологической пульсации нет. Верхушечный толчок пальпируется в V-ом межреберье слева на 1,5 см кнутри от срединно-ключичной линии, площадь его 1,5 см, умеренной высоты и резистентности. Сердечный толчок не выявляется. Симптом кошачьего мурлыканья отсутствует. При аускультации тоны сердца приглушены, ритмичные, ЧСС- 78 в 1 мин. Патологические шумы не выслушиваются. При исследовании сосудов определяется мягкая, эластичная, ровная, плоская стенка лучевой артерии. Пульс — одинаковый на обеих руках, ритмичный, частота-78 пульсовых волны в минуту, удовлетворительного напряжения, полный. Дефицита пульса нет. Артериальное давление: 150/90 мм рт.ст на обеих руках.
Система органов пищеварения
Слизистые щек, губ, твердого неба розового цвета. Язык обычных размеров, розовый, влажный, чистый, сосочки сохранены; обложенность, трещины, опухоли, язвы отсутствуют. Полость рта санирована удовлетворительно. Миндалины не изменены, гнойные пробки и налет отсутствуют. Слизистая сухая, гладкая. Живот округлой формы, симметричный, брюшная стенка равномерно участвует в акте дыхания. При поверхностной пальпации живот мягкий. Глубокая пальпация затруднена. Перистальтика кишечника обычная. Перитонеальные симптомы отсутствуют. Пальпация безболезненная. Стул 1 раз в сутки, не нарушен. Печень без-болезненна, границы в норме. Селезёнка не увеличена.
Мочеполовая система
Мочеиспускание свободное, безболезненное. Почки не пальпируются. Точки почек и мочевыводящих путей безболезненны. Симптом поколачивания отрицательный с обеих сторон. Диурез достаточный.
6. ПРЕДВАРИТЕЛЬНЫЙ ДИАГНОЗ
На основании жалоб больной (общая слабость, головокружение, одышка при физических нагрузках и в покое, повышенную утомляемость); анамнеза (больна с августа 2013, 2 раза проходила лечение в стационаре); данных объективного исследования (бледность кожных покровов) можно выставить предварительный диагноз – Аутоиммунная гемолитическая анемия.
7. ПЛАН ОБСЛЕДОВАНИЯ БОЛЬНОГО
ОАК, ОАМ, БАК
Иммунологический анализ
Кровь на RW, Hbs, анти HCV
УЗИ ОБП
ЭКГ
8. РЕЗУЛЬТАТЫ ЛАБОРАТОРНЫХ И ИНСТРУМЕНТАЛЬНЫХ
МЕТОДОВ ИССЛЕДОВАНИЯ
1. Общий анализ крови (15.11.13)
Гемоглобин: 81 г/л (ниже нормы)
Эритроциты: 2,33х1012 /л (ниже нормы)
СОЭ:29 мм в ч. (выше нормы)
Лейкоциты: 5.9 х109 /л
-миелоциты: 1%
-палочкоядерные: 9%
-сегментоядерные: 46%
-лимфоциты: 40%
-моноциты:4%
Токсическая зернистость нейтрофилов ++
Тромбоциты: 283х109/л
Общий анализ крови (18.11.13)
Гемоглобин: 113 г/л (ниже нормы)
Эритроциты: 3,2 х1012 /л (ниже нормы)
СОЭ:21 мм в ч. (выше нормы)
Лейкоциты: 6,3 х109 /л
-миелоциты: 1%
-юные: 1%
-палочкоядерные: 3%
-сегментоядерные: 40%
-лимфоциты: 41%
-моноциты:12%
-базофилы: 1%
-эозинофилы: 2%
Тромбоциты: 295х109/л
2. Биохимический анализ крови (15.11.13)
АЛТ: 32 Е/л
АСТ: 25 Е/л
Глюкоза: 6,1 ммоль/л
Билирубин: общий – 17,7 мкмоль/л, прямой -2,5 мкмоль/л
Щелочная фосфатаза: 38 Ед/л
Общий белок: 59 г/л
Мочевина: 9,5
Креатинин: 0,098
ЛДГ: 639 ед/л
Биохимический анализ крови (18.11.13)
АЛТ: 47 Е/л
АСТ: 41 Е/л
Глюкоза: 5,3 ммоль/л
Билирубин: общий – 10,7 мкмоль/л
Общий белок: 65 г/л
Мочевина: 5,7
Креатинин: 0,062
ЛДГ: 639 ед/л
3. Общий анализ мочи (15.11.13)
Цвет: соломенно-желтый
Реакция: кислая
Удельный вес: 1,011 г/мл
Прозрачность: мутная
Белок: нет
Сахар: нет
Эпителий (плоский): 1-2 в поле зрения
Лейкоциты: 3-4 в поле зрения
Ураты ++
4. Иммунологический анализ(16.11.2013)
Увеличение циркулирующих иммунных комплексов. Относительное сни-жение показателей Т-клеточного иммунитета.
9. КЛИНИЧЕСКИЙ ДИАГНОЗ И ЕГО ОБОСНОВАНИЕ
На основании жалоб больной (общая слабость, головокружение, одышка при физических нагрузках и в покое, повышенную утомляемость); анамнеза (больна с августа 2013, 2 раза проходила лечение в стационаре); данных объективного исследования (бледность кожных покровов); данных лабораторных и инструментальных исследований (низкий уровень гемоглобина, эритроцитов; миелоциты,юные, токсическая зернистость нейтрофилов в ОАК) выставляется окончательный диагноз – Аутоиммунная гемолитическая анемия, ср. степени тяжести, протекающая с мегалобластоидностью костного мозга. Кардиомиопатия смешанного генеза Н 1. АГ 2ст риск 4.
10. ДИФФЕРЕНЦИАЛЬНЫЙ ДИАГНОЗ
Дифференциальная диагностика проводится с:
• А. Мегалобластными анемиями (заболевания, характеризующиеся изменениями морфологии клеток к.м. вследствие нарушения синтеза ДНК. Более 90 % — В-12 и фолиево-дефицитные анемии).
После начала терапии витамином В-12 или фолиевой кислотой в анализе крови выявляется ретикулярный криз на 5-7 сутки и повышение показателей красной крови, что нехарактерно для больных рефрактерной анемией. Изменения кариотипа клеток костного мозга не встречаются при мегалобластных анемиях.
• Б. Апластической анемией.
Апластическая анемия может быть врождённой, приобретённой и идиопатиче-ской. Врождённая апластическая анемия — анемия Фанкони сочетается с другими генетическими аномалиями (кожная пигментация, гипоплазия по-чек, микроцефалия), приобретённая связана с действием химических и физических агентов, инфекциями, нарушениями обмена веществ.
Для АА нехарактерны изменение кариотипа, гиперклеточный костный мозг.
• В. Анемии при ХПН.
• В. Анемии при хроническом активном гепатите характерно выявление маркеров вирусных инфекций, гепатоспленомегалия, клиническая картина хр. гепатита, изменения биохимических показателей крови (метаболизма билирубина, функции печени).
План лечения:
Режим П, Стол Г., поливитамины.
1. Переливание Эр. Массы
2. Глюкокортикоиды — Преднизолон в суточной дозе 60—80 мг
3. Иммунодепрессанты – Метотрексат, циклофосфамид
4. Дезинтоксикационная терапия.
20.11. Жалобы на общую слабость, головокружение, одышка при физических нагрузках, повышенную утомляемость.
Состояние больной удовлетворительное. Сознание ясное. Кожный покров и видимые слизистые оболочки бледного цвета, сухие, чистые, тургор удовлетворительный. Тоны сердца ритмичные, приглушены. ЧСС – 58 уд. в мин. АД – 140/80 мм.рт.ст.. Дыхание везикулярное. ЧД – 16 в мин. Живот мягкий, безболезненный. Стул в норме. Мочеиспускание свободное, безболезненное.
Рекомендовано: Лечение продолжить
21.11. Жалобы на общую слабость, головокружение, одышка при физических нагрузках, повышенную утомляемость.
Состояние больной удовлетворительное. Сознание ясное. Кожный покров и водимые слизистые без особенностей. Тоны сердца ритмичные, приглушены. ЧСС – 64 в мин. АД – 150/85 мм.рт.ст. Дыхание везикулярное. ЧД – 17 в мин. Живот мягкий, безболезненный. Стул был. Удовлетворительных свойств. Мочеиспускание свободное, безболезненное.
Лечение продолжать.
13. ЭПИКРИЗ
Больная ___________, 02.10.1953 г. р., пенсионерка, поступила 14.11.13. в гематологическое отделение УЗ ВОКБ в плановом порядке для лечения с жалобами на общую слабость, головокружение, одышка при физических нагрузках и в покое, повышенную утомляемость. Болеет с августа 2013. Два раза проходила лечение в стационаре.
Объективное исследование- бледность кожных покровов. Лабораторные данные – снижен уровень гемоглобина, эритроцитов; наличие юных, миелоцитов, токсической зернистости нейтрофилов.
На основании комплексного клинико-лабороторного обследования выставлен диагноз: Аутоиммунная гемолитическая анемия, ср. степени тяжести, протекающая с мегалобластоидностью костного мозга. Кардиомиопатия смешанного генеза Н 1. АГ 2ст риск 4.
Пациентка находится в стационаре. Отмечается положительная динамика.
Рекомендовано – лечение продолжить.
источник
Анамнез жизни и заболевания пациентки. Проведение обследования кожных покровов и систем организма больного. Клинический и биохимический анализ крови. Обоснование дифференциального диагноза «гипохромная анемия тяжелой степени», назначение лечения.
Студенты, аспиранты, молодые ученые, использующие базу знаний в своей учебе и работе, будут вам очень благодарны.
Размещено на http://www.allbest.ru/
Диагноз основной: Железодефицитная анемия тяжелой степени.
Диагноз сопутствующий: Хронический нодулярный гастрит.
Юля, 15 лет, поступила на гематологическое отделение ДГБ№1 06.02.2013 с жалобами на бледность кожи, головокружения, ощущения сердцебиения, быструю утомляемость.
Анамнез болезни: Бледность кожи, заеды в углах рта отмечает в течение длительного времени. С лета 2012 года- головокружения, сдали анализ крови, в котором HGB 74г/л, были назначены витамины, без эффекта. В динамике появилось извращение вкуса (хочет мела), сохранялись эпизоды головокружения, сердцебиения. 4.02.13 вновь сдали анализ крови- HGB 54г/л, RBC 3,37*1012/л, госпитализирована.
С 8 лет ограничивала мясные продукты (со слов девочки — не любит). В питании — молочно-растительные продукты, редко курица. В 2011-2012г с целью изменения веса ограничивала объем питания. Тогда же — аменорея в течение 6 месяцев, проходила консультации психолога. В 2012 году — ростовой скачек- +10см. В настоящий момент вес 54 кг, рост 170см (ИМТ=19). Менструации нормализовались.
Черный стул, кровь в стуле отрицает. Периодически — повышенное выпадение волос. Получает курсы поливитаминов.
Дополнительные занятия спортом 5-6раз в неделю (фитнес, бассейн). В течение полугода — повышенная утомляемость, сонливость, снижение толерантности к физ. нагрузке.
Анамнез жизни: Девочка родилась в срок, в физическом и психомоторном развитии от сверстников не отставала. Привита в соответствии с календарем прививок, детскими инфекциями не болела. Аллергия пищевая (цитрусовые). Травм, операций, трансфузий не было. Менструации с 12 лет, на данный момент регулярные, обильные.
Объективный осмотр (на момент курации-08.02.13)
Состояние средней степени тяжести. Самочувствие удовлетворительное. Фон настроения снижен.
Кожа чистая, бледная, «тени» под глазами. Влажность кожи умеренная, эластичность сохранена. Волосистая часть головы без облысений, волосы тусклые, ломкие. Ногти овальной формы, ломкие. Симптом «щипка», «жгута» отрицательный. Дермографизм белый не разлитой, время появления — сразу, исчезает через -10 сек. Видимые слизистые оболочки бледно-розового цвета, без изменений. Язык влажный, гипотрофия сосочков. Хейлит.
Подкожно-жировая клетчатка развита умеренно, распределена равномерно.
Толщина жирового слоя на уровне пупка, под реберной дугой, под углами лопаток, на плечах и бедрах — 1,5 см. Тургор сохранен. Уплотнения, отеки отсутствуют. Пальпируются подчелюстные, подмышечные лимфоузлы до 5 мм в диаметре, единичные, с гладкой поверхностью, безболезненные, подвижные, не спаянные с окружающими тканями. Мышечная система: мышцы развиты умеренно, симметрично; при пальпации мышцы безболезненны; тонус мышц при пассивном сгибании и разгибании на симметричных участках одинаковый.
Исследование органов кровообращения.
В области сердца деформаций грудной клетки нет, патологической пульсации сонных артерий, набухание яремных вен не выявлено. Верхушечный толчок локализуется в V межреберье на 0,5 см кнутри от среднеключичной линии, площадь 2*2 см, умеренной силы. Патологическая пульсация в области сердца, систолическое и пресистолическое дрожание не выявлено. Пульс 96уд/мин симметричный, ритмичный, мягкий, малого наполнения. Аускультативно: тоны сердца ритмичные, ослабление I тона над верхушкой, систолический шум над верхушкой. АД 100/60 мм рт ст. Заключение: отмечается умеренная тахикардия, ослабление I тона и систолический шум над верхушкой.
Исследование органов дыхания
Носовое дыхание свободно. Вспомогательные мышцы в акте дыхания не участвуют. Отмечается параорбикулярный цианоз. Экскурсия грудной клетки в полном объеме. Грудная клетка цилиндрической формы, нормостеническая, симметричная, обе половины активно участвуют в акте дыхания. Тип дыхания — смешанный, глубина средняя, частота — 20 в минуту. При пальпации грудная клетка безболезненна, эластичная, голосовое дрожание на симметричных участках проводится одинаково. При сравнительной перкуссии над всей легочной поверхностью выслушивается ясный лёгочный звук. При аускультации лёгких на симметричных участках определяется везикулярное дыхание. Бронхофония не изменена. Патологические дыхательные шумы не выслушиваются. Заключение: патологические изменения не выявлены. Исследование органов пищеварения Живот обычных размеров, симметричный, не изменен. Рубцы и грыжевые выпячивания не выявлены. Венозная сеть не выражена. Видимая перистальтика отсутствует. При поверхностной пальпации живот мягкий, безболезненный. Симптомы раздражения брюшины отрицательные. Печень выступает на 1 см из-под края реберной дуги, безболезненная, край ее ровный, эластичный, закруглен. Размеры печени по Курлову 11-10-8 см. Симптомы Кера, Мерфи, Мюсси, Ортнера отрицательные. Селезенка не пальпируется. Заключение: На момент курации патологических изменений со стороны ЖКТ не выявлено.
Периферические отеки при осмотре не выявлены. Почки не пальпируются. Мочеточниковые точки безболезненные. Мочевой пузырь не выступает над лоном, не пальпируется. Симптом поколачивания по поясничной области отрицательный с обеих сторон. Мочеиспускание безболезненное, не нарушено. Заключение: со стороны мочевыделительной системы патологии не выявлено.
На основании жалоб на выраженную бледность кожных покровов, быструю утомляемость, слабость, на ощущения сердцебиения, головокружение; на основании особенностей пищевого поведения девочки (не употребляет в пищу мясо); на основании факта инверсии вкуса (мел); на основании данных объективного осмотра- выраженная бледность кожи, «тени» под глазами, хейлит, ломкость, тусклость волос и ногтей; умеренная тахикардия, ослабление 1тона и систолический шум над верхушкой; на основании данных лабораторных исследований (проведены амбулаторно 4.02.13) — в гемограмме — HGB 54г/л, RBC 3,37*1012/л; уровень железа в сыворотке- 0,30мкмоль/л, концентрация ферритина — 5,92мкг/л, можно поставить предварительный диагноз: Железодефицитная анемия тяжелой степени.
План дальнейшего исследования:
Поиск источников оккультного кровотечения — ФГДС, ФКС, анализ кала на скрытую кровь.
Биохимический анализ крови: исследование сывороточного железа, общей железосвязывающей способности сыворотки крови, латентная железосвязывающая способности сыворотки крови, билирубин, АсАт.
Определения вит. В12, фолиевой кислоты
Данные лабораторных и инструментальных исследований:
источник
Гемолитическая анемия – патология эритроцитов, отличительным признаком которой является ускоренное разрушение красных кровяных телец с высвобождением повышенного количества непрямого билирубина. Для гемолитических анемий типично сочетание анемического синдрома, желтухи и увеличения размеров селезенки. В процессе диагностики исследуется общий анализ крови, уровень билирубина, анализ кала и мочи, УЗИ органов брюшной полости; проводится биопсия костного мозга, иммунологические исследования. В качестве методов лечения гемолитической анемии используется медикаментозная, гемотрансфузионная терапия; при гиперспленизме показана спленэктомия.
Гемолитическая анемия — анемия, обусловленная нарушением жизненного цикла эритроцитов, а именно преобладанием процессов их разрушения (эритроцитолиза) над образованием и созреванием (эритропоэзом). Группа гемолитических анемий очень обширна. Их распространенность неодинакова в различных географических широтах и возрастных группах; в среднем патология встречается у 1% населения. Среди прочих видов анемий на долю гемолитических приходится 11%. При гемолитической анемии жизненный цикл эритроцитов укорочен и их распад (гемолиз) происходит раньше времени (через 14-21 день вместо 100-120 суток в норме). При этом разрушение эритроцитов может происходить непосредственно в сосудистом русле (внутрисосудистый гемолиз) или в селезенке, печени, костном мозге (внесосудистый гемолиз).
В гематологии гемолитические анемии подразделяются на две большие группы: врожденные (наследственные) и приобретенные.
Наследственные гемолитические анемии включают следующие формы:
- эритроцитарные мембранопатии (микросфероцитоз – болезнь Минковского-Шоффара, овалоцитоз, акантоцитоз) – гемолитические анемии, обусловлены структурными аномалиями мембран эритроцитов
- ферментопении (энзимопении) – гемолитические анемии, вызванные дефицитом тех или иных ферментов (глюкозо-6-фосфатдегидрогеназы, пируваткиназы и др.)
- гемоглобинопатии — гемолитические анемии, связанные с качественными нарушениями структуры гемоглобина или изменением соотношения его нормальных форм (талассемия, серповидно-клеточная анемия).
Приобретенные гемолитические анемии подразделяются на:
- мембранопатии приобретенные (пароксизмальная ночная гемоглобинурия – б-нь Маркиафавы-Микели, шпороклеточная анемия)
- иммунные (ауто- и изоиммунные анемии) – обусловлены воздействием антител
- токсические – гемолитические анемии, обусловленные воздействием химических веществ, биологических ядов, бактериальных токсинов
- гемолитические анемии, вызванные механическим повреждением структуры эритроцитов (тромбоцитопеническая пурпура, маршевая гемоглобинурия)
Патогенетическую основу наследственных гемолитических анемий составляют генетические дефекты мембран эритроцитов, их ферментных систем либо структуры гемоглобина. Данные предпосылки обусловливают морфо-функциональную неполноценность эритроцитов и их повышенное разрушение. Гемолиз эритроцитов при приобретенных анемиях наступает под влиянием внутренних факторов или факторов окружающей среды.
Развитию иммунных гемолитических анемий могут способствовать посттрансфузионные реакции, профилактическая вакцинация, гемолитическая болезнь плода, прием определенных лекарств (противомалярийных препаратов, сульфаниламидов, производных нитрофуранового ряда, анальгетиков). Аутоиммунные реакции с образованием антител, агглютинирующих эритроциты, возможны при гемобластозах (остром лейкозе, хроническом лимфолейкозе, лимфогранулематозе, миеломной болезни), аутоиммунной патологии (СКВ, неспецифическом язвенном колите), инфекционных заболеваниях (инфекционном мононуклеозе, токсоплазмозе, сифилисе, вирусной пневмонии).
В ряде случаев острому внутрисосудистому гемолизу предшествует отравление мышьяковистыми соединениями, тяжелыми металлами, уксусной кислотой, грибными ядами, алкоголем и др. Механическое повреждение и гемолиз эритроцитов может наблюдаться при тяжелых физических нагрузках (длительной ходьбе, беге, лыжном переходе), при ДВС-синдроме, малярии, злокачественной артериальной гипертензии, протезировании клапанов сердца и сосудов, проведении гипербарической оксигенации, сепсисе, обширных ожогах. В этих случаях под действием тех или иных факторов происходит травматизация и разрыв мембран изначально полноценных эритроцитов.
Центральным звеном патогенеза гемолитических анемий является повышенное разрушение эритроцитов в органах ретикулоэндотелиальной системы (селезенке, печени, костном мозге, лимфатических узлах) или непосредственно в сосудистом русле. Эти процессы сопровождаются развитием анемического и желтушного синдромов (так называемой «бледной желтухой»). Возможно интенсивное окрашивание кала и мочи, увеличение селезенки и печени.
Наиболее распространенной формой данной группы гемолитических анемий является микросфероцитоз, или болезнь Минковского-Шоффара. Наследуется по аутосомно-доминантному типу; обычно прослеживается у нескольких представителей семьи. Дефектность эритроцитов обусловлена дефицитом в мембране актомиозиноподобного белка и липидов, что приводит к изменению формы и диаметра эритроцитов, их массивному и преждевременному гемолизу в селезенке.
Манифестация микросфероцитарной гемолитической анемии возможна в любом возрасте (в младенчестве, юношестве, старости), однако обычно проявления возникают у детей старшего возраста и подростков. Тяжесть заболевания варьирует от субклинического течения до тяжелых форм, характеризующихся часто повторяющимися гемолитическими кризами. В момент криза нарастает температура тела, головокружение, слабость; возникают боли в животе и рвота.
Основным признаком микросфероцитарной гемолитической анемии служит желтуха различной степени интенсивности. Вследствие высокого содержания стеркобилина кал становится интенсивно окрашенным в темно-коричневый цвет. У пациентов с болезнь Минковского-Шоффара наблюдается склонность к образованию камней в желчном пузыре, поэтому часто развиваются признаки обострения калькулезного холецистита, возникают приступы желчной колики, а при закупорке холедоха конкрементом — обтурационная желтуха. При микросфероцитарной гемолитической анемии во всех случаях увеличена селезенка, а у половины пациентов – еще и печень.
Кроме наследственной микросфероцитарной гемолитической анемии, у детей часто встречаются другие врожденные дисплазии: башенный череп, косоглазие, седловидная деформация носа, аномалии прикуса, готическое нёбо, полидактилия или брадидактилия и пр. Пациенты среднего и пожилого возраста страдают трофическими язвами голени, которые возникают в результате гемолиза эритроцитов в капиллярах конечностей и плохо поддаются лечению.
Энзимопенические гемолитические анемии связаны с недостатком определенных ферментов эритроцитов (чаще — Г-6-ФД, глутатион-зависимых ферментов, пируваткиназы и др). Гемолитическая анемия может впервые заявлять о себе после перенесенного интеркуррентного заболевания или приема медикаментов (салицилатов, сульфаниламидов, нитрофуранов). Обычно заболевание имеет ровное течение; типична «бледная желтуха», умеренная гепатоспленомегалия, сердечные шумы. В тяжелых случаях развивается ярко выраженная картина гемолитического криза (слабость, рвота, одышка, сердцебиение, коллаптоидное состояние). В связи с внутрисосудистым гемолизом эритроцитов и выделением гемосидерина с мочой последняя приобретает темный (иногда черный) цвет.
Особенностям клинического течения гемоглобинопатий — талассемии и серповидно-клеточной анемии посвящены самостоятельные обзоры.
Среди различных вариантов приобретенных гемолитических анемий чаще других встречаются аутоиммунные анемии. Для них общим пусковым фактором выступает образование антител к антигенам собственных эритроцитов. Гемолиз эритроцитов может носить как внутрисосудистый, так и внутриклеточный характер.
Гемолитический криз при аутоиммунной анемии развивается остро и внезапно. Он протекает с лихорадкой, резкой слабостью, головокружением, сердцебиением, одышкой, болями в эпигастрии и пояснице. Иногда острым проявлениям предшествуют предвестники в виде субфебрилитета и артралгий. В период криза стремительно нарастает желтуха, не сопровождающаяся кожным зудом, увеличивается печень и селезенка.
При некоторых формах аутоиммунных гемолитических анемий больные плохо переносят холод; в условиях низких температур у них может развиваться синдром Рейно, крапивница, гемоглобинурия. Вследствие недостаточности кровообращения в мелких сосудах возможны осложнения в виде гангрены пальцев ног и рук.
Токсические гемолитические анемии протекают с прогрессирующей слабостью, болями в правом подреберье и поясничной области, рвотой, гемоглобинурией, высокой температурой тела. Со 2-3 суток присоединяется желтуха и билирубинемия; на 3-5 сутки возникает печеночная и почечная недостаточность, признаками которых служат гепатомегалия, ферментемия, азотемия, анурия.
Отдельные виды приобретенных гемолитических анемий рассмотрены в соответствующих статьях: «Гемоглобинурия» и «Тромбоцитопеническая пурпура», «Гемолитическая болезнь плода».
Определение формы гемолитической анемии на основе анализа причин, симптоматики и объективных данных относится к компетенции гематолога. При первичной беседе выясняется семейный анамнез, частота и тяжесть протекания гемолитических кризов. В процессе осмотра оценивается окраска кожных покровов, склер и видимых слизистых, производится пальпация живота для оценки величины печени и селезенки. Сплено- и гепатомегалия подтверждается при проведении УЗИ печени и селезенки.
Изменения в гемограмме характеризуются нормо- или гипохромной анемией, лейкопенией, тромбоцитопенией, ретикулоцитозом, ускорением СОЭ. При аутоиммунных гемолитических анемиях большое диагностическое значение имеет положительная проба Кумбса. В биохимических пробах крови определяется гипербилирубинемия (увеличение фракции непрямого билирубина), увеличение активности лактатдегидрогеназы. Исследование мочи выявляет протеинурию, уробилинурию, гемосидеринурию, гемоглобинурию. В копрограмме повышено содержание стеркобилина. Исследование пунктата костного мозга обнаруживает гиперплазию эритроидного ростка.
В процессе дифференциальной диагностики исключаются гепатиты, цирроз печени, портальная гипертензия, гепатолиенальный синдром, гемобластозы.
Различные формы гемолитической анемии имеют свои особенности и подходы к лечению. При всех формах приобретенной гемолитической анемии необходимо позаботиться об устранении влияния гемолизирующих факторов. Во время гемолитических кризов больным необходимы инфузии растворов, плазмы крови; витаминотерапия, по необходимости – гормоно- и антибиотикотерапия. При микросфероцитозе единственно эффективным методом, приводящим к 100 % прекращению гемолиза, является спленэктомия.
При аутоиммунной гемолитической анемии показана терапия глюкокортикоидными гормонами (преднизолоном), сокращающая или прекращающая гемолиз. В некоторых случаях требуемый эффект достигается назначением иммунодепрессантов (азатиоприна, 6-меркаптопурина, хлорамбуцила), противомалярийных препаратов (хлорохина). При резистентных к медикаментозной терапии формах аутоиммунной гемолитической анемии выполняется спленэктомия.
Лечение гемоглобинурии предполагает переливание отмытых эритроцитов, плазмозаменителей, назначение антикоагулянтов и антиагрегантов. Развитие токсической гемолитической анемии диктует необходимость проведения интенсивной терапии: дезинтоксикации, форсированного диуреза, гемодиализа, по показаниям – введение антидотов. При развитии почечной недостаточности прогноз неблагоприятен.
источник
Среди приобретенных экзоэритроцитных гемолитических анемий аутоиммунные гемолитические анемии (АИГА) наиболее часто встречается в клинической практике, даже чаще микросфероцитной гемолитической анемии и талассемии.
По механизму развития они рассматриваются как типично аутоиммунные заболевания, при этом гемолиз — характерное поражение — происходит под непосредственным воздействием антител на оболочку эритроцита. По классификации Gell и Coombs (Берчану) этот механизм поражения отнесен к типу 2.
Проведенные после 1960 г. исследования уточнили механизм расплавления и природу антител, однако, также как и при других аутоиммунных болезнях, еще не подтверждена гипотеза о развитии состояния сельф-сенсибилизации, нарушения иммунной переносимости и появления иммунной реакции антителом в отношении собственных структур — антигены эритроцита.
В генуинных формах заболевания аутоиммунный патогенез нелегко объяснить, тем не менее знание вторичных форм, с определенным патогенезом, подсказывает новые патогенетические гипотезы и в отношении генуинных форм.
В существующих двух монографиях (Месробяну и Берчану) дано общее описание этих патогенетических механизмов, как в специальных главах с аутоиммунной гемолитической анемией (АИГА), так и в главах общей патологии явлений аутоиммунизации.
Современные знания о аутоиммунной гемолитической анемии (АИГА) результат продолжительных наблюдений и исследований, началом которых являются описанные во Франции случаи аутоиммунной гемолитической анемии (АИГА), рассматриваемые как спленомегалическая хроническая инфекционная желтуха (Hayem), а позже как «приобретенная гемолитическая желтуха», отличающаяся от врожденной выявлением Видали, Абрами и Брюле аутоагглютинин — характерных серологических факторов.
В течение того же периода, так же во Франции, Chauffatd и его сотрудники Troisier и Vincent описали гемолизиновую гемоглобинурию и желтуху. В своей монографии о гемолитической анемии Dacie дает полное описание эволюции знаний и в частности серологических методов, уточнивших диагностику всех существующих в то время форм аутоиммунной гемолитической анемии (АИГА).
Лишь спустя 20 лет после первых, проведенных во Франции наблюдений, были описаны Brill и Ledderer новые формы острого аутоиммунного гемолиза, но без серологической ссылки. Позже Dyke и Young выделили формы макроцитной гемолитической анемии — различные с гематологической точки зрения и по реакции на удаление селезенки — и сфероцитной анемии, однако не опирающиеся на серологическое исследование.
В 1940 г. Dameshek и Schwartz внесли новый серологический вклад описанием аномальных гемолизинов при приобретенной острой гемолитической анемии.
Крутой поворот произошел в 1945 году после уточнения Coombs, Mourand и Rice теста агглютинации антиглобулиновой сывороткой. При этом были вскрыты «неполные» антитела, по началу в случаях изоиммунизации за счет несовместимости в Rh-системе. До 1950 г. было установлено, что прямая реакция Кумбса дает положительный результат в приобретенных формах гемолитической анемии, отличающихся по своему течению от микросфероцитной гемолитической анемии (Loutit и Mollison). Так, были выделены генуинные приобретенные гемолитические анемии с неполными антителами. В 1947 г. Morton и Pickles внедрили трипсинизацию, как метод сенсибилизации реакции агглютинации посредством аутоантител.
После 1950 г., в связи с развитием концепции аутоиммунного заболевания, гемолитическая анемия с неполными антителами была отнесена к группе аутоиммунных болезней, при этом развитие новых методов, применяемых этой группе болезней способствует дифференциации всех видов аутоиммунной гемолитической анемии (АИГА) с уточнением биологической и биохимической характеристикой природой аутоантител.
Вскрытие аутоиммунной гемолитической анемии (АИГА) у животных, в частности у мышей NZB (Бильшовский и сотр.) освещает под новым углом патогенетическую взаиосвязь гемолитических аутоантител и лимфопролиферативных заболеваний. Даются ценные сведения о аутоиммунной гемолитической анемии (АИГА) при хронической лимфоцитной анемии (Pisciotta), рассматриваемой как злокачественное заболевание иммуной лимфоидной системы, способствующее утрате врожденной иммунной переносимости и развитию аутоантител (Burnet, Dameshek, Mackay и Burnet).
Проведенные после 1965 г. исследования уточнили диагностику отдельных форм аутоиммунной гемолитической анемии (АИГА) и описали некоторые частные виды, уточнение которых осуществляется также специфической иммуносерологической реакцией.
В настоящее время в нашем распоряжении имеется достаточное число клинических и биологических параметров, делаюших возможной классификацию аутоиммунной гемолитической анемии (АИГА) по научной номенклатуре. Считаем нецелесообразным применение прежней номенклатуры классических авторов, поскольку отграниченные ими формы гемолитической анемии крайне полиморфны, и большинство их не основывается на серологическом исследовании. Так, на современной стадии, представляют лишь историческое значение термины приобретенной гемолитической анемии вида Hayem-Vidal, Dyke-Young, Ledderer-Brill и пр.
Сохраняется термин острой гемолтическоий анемии с двухфазными агглютининами Donath-Landsteiner, описанной в 1904 г., но в настоящее время известной больше под названием холодовой пароксизмальной анемии; также название болезни Marchiafava-Michelli заменено термином ночной пароксизмальной гемоглобинурии.
Статистика частоты генуинной аутоиммунной гемолитической анемии в Бухарестском Институте внутренней медицины (по Георгиу и сотр.) 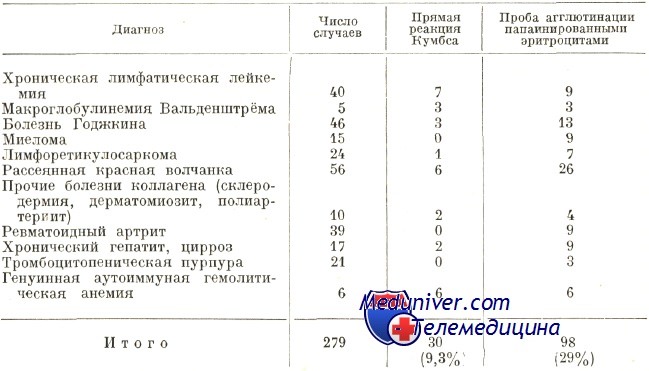
Современная классификация основывается на внесенный иммуносерологической методологией вклад в дело уточнения механизмов аутоиммунного гемолиза с помощью полных и неполных антител (Dacie, Van Loghen, Worlledge, Dacie и Worlledge, Pirofsky, Leddy и сотр.), которые действуют при тепловых и Холодовых реакциях в процессе диагностирования. С клинической точки зрения понятия полных или неполных холодовых или тепловых антител в настоящее время достаточны для определения серологической характеристики той или иной АИГА.
Однако исследование специфической природы гемолитических аутоантител уточнило, что их наибольшая часть специфична для определенных групп крови. В то же время поскольку речь идет о иммуноглобулинах, они принадлежат основным классам IgG, IgM или IgA и в реакциях с антигенами связывают или не связывают комплемент (Werner и Vos, Worlledge, Leddy и сотр., Dacie и Worlledge).
С практической точки зрения постановкой диагноза аутоиммунной гемолитической анемии (АИГА) преследуется выделение двух основных групп — аутоиммунной гемолитической анемии с тепловыми антителами и аутоиммунной гемолитической анемии с холодовыми антителами. В обеих группах антитела бывают полными и неполными, однако, в принципе, в группе тепловых антител преобладают неполные. Следует отметить, что, при аутоиммунной гемолитической анемии с холодовыми антителами у данного больного в течение острого периода наблюдается одновременное наличие обоих видов — полных и неполных —, в то время как на хронической стадии — в основном неполные антитела.
В начале аутоиммунной гемолитической анемии были описаны как генуинные заболевания, тем не менее во всех последних статистиках первичные генуинные формы реже наблюдаются, в то время как вторичные — преобладают. В таблице на 279 случаях, находящихся под наблюдением Института внутренней медицины (Георгиу и сотр.), можно проследить распределение вторичных форм по отдельным группам заболеваний и невысокий показатель частоты генуинных форм, что противоречит статистике Worlledge. В иных статистиках значатся вторичные аутоиммунной гемолитической анемии при неоплазии, в основном яичников (Van Loghen), также при затяжных хронических инфекциях.
По Dacie распределение равномерное, по 50% на генуинную и соответственно вторичную формы; Worlledge говорит о 80% генуинных форм, вто время как Dausset и Colombani о 30%, a Pirofsky на 234 случаях отметил лишь 18,8%. Известно, что генуинные формы рассматриваются как таковые, поскольку отсутствуют признаки иных первичных или вторичных заболеваний. Во многих случаях после определенного периода эволюции генуинной АИГА появляются симптомы лимфопролиферативной или другой комплексной аутоиммунной болезни, такой как, РКВ, склеродермия, ЭХП (РСЕ). Однако, в отдельных случаях, генуинная форма диагностируется как аутоиммунный гемолиз лишь при использовании всего набора иммунологических тестов.
Недавно составленная нами в гематологической клинике больницы Фундень статистика на 70 случаях свидетельствует о 24 генуинных (34,2%) и 46 вторичных (67,8%) формах. В злокачественных заболеваниях крови наблюдается преобладание вторичных форм. Отнесение случаев к первичной или вторичной категории возможно зависит от профиля медицинского учреждения, под наблюдением которого находились больные. Это может объяснить высокий показатель численности форм вторичного заболевания, особенно при РКВ и ХЛЛ по более старым статистикам Института внутренней медицины.
Тяжелые формы аутоиммунной гемолитической анемии определяются характеризующим их видом антитела — холодовым и тепловым.
По нашему мнению формы с холодовыми антителами не выделяются в особую группу (Issit) и следовательно оба вида составляют одну лишь единицу аутоиммунной гемолитической анемии с серологической и клинической разницами (Brown, Берчану).
источник
Этиология и патогенез. В патогенезе приобретенной аутоиммунной гемолитической анемии (anaemia haemolytica chronica) основное значение придается иммунопатологическим сдвигам, заключающимся в выработке антител к собственным эритроцитам — аутоагглютининов. Спровоцировать эту выработку антител могут некоторые острые инфекции, интоксикации, в том числе и лекарственные, особо тяжелые формы злокачественных лимфом и коллагенозов, а также другие факторы. Эти антитела относятся к фракции иммуноглобулинов и являются неполными, «слабыми», антителами: фиксируясь на поверхности эритроцитов в кровяном русле, они не вызывают агглютинации, но «блокируют» эритроциты, облегчая их оседание в депо ретикулогистиоцитарной системы (прежде всего в венозных синусах селезенки), а затем захват и разрушение макрофагами.
Иногда аутоиммунная гемолитическая анемия связана с возникновением Холодовых которые вместе с комплементом фиксируются на эритроцитах. Их действие проявляется в периферических участках тела (кончики пальцев, уши) при переохлаждении. У некоторых больных, помимо аутоагглютининов, выявляются также аутогемолизины; в этих случаях заболевание может протекать с признаками не тольконо и внутрисосудистого гемолиза.
Клиническая картина. Болезнь начинается или постепенно, незаметно, или остро, с гемолитического криза. Основными являются жалобы на слабость, головокружение, утомляемость, незначительное повышение температуры тела. Все эти явления резко усиливаются в период гемолитических кризов. Кожного зуда не бывает. Кожа больных бледная, с несколько желтушным оттенком. Давление на грудину и перкуссия по ней болезненны. Палъпаторно определяется увеличенная и уплотненная селезенка, отмечается небольшое увеличение печени.
В крови снижено содержание эритроцитов и гемоглобина при нормальном цветовом показателе. Эритроциты неодинаковой формы, размеров и окраски (пойкилоцитоз, анизоцитоз, анизохромия); средний размер их несколько меньше, чем в норме (микроцитоз). В отличие от врожденной гемолитической анемии при приобретенной, как и у здоровых лиц, эритроциты в центре окрашены слабее, чем по краям, что зависит
от их формы (планоциты). Много ретикулоцитов, особенно выражен ретикулоцитоз при значительной анемизации и в период после гемолитических кризов. Осмотическая резистентность эритроцитов существенно не изменена. Сыворотка крови больных имеет желтоватую окраску — исследование крови подтверждает повышение содержания свободного билирубина, от которого и зависит ее цвет. Определяется также гипергаммаглобулинемия, повышено содержание железа сыворотки, которое высвобождается в большом количестве при гемолизе эритроцитов. Вследствие повышенного выделения билирубина желчь, полученная при дуоденальном зондировании, имеет очень темную окраску. Моча и кал больного также окрашены темнее, чем у здоровых лиц, суточное выделение стеркобилина с калом и уробилина с мочой увеличено. Пунктат костного мозга свидетельствует о более или менее значительном усилении эритропоэза.
В крови больных аутоиммунной гемолитической анемией выявляются антитела, как фиксированные на поверхности эритроцитов (блокирующие), так и находящиеся в свободном состоянии в плазме (конглютинины). Для их обнаружения применяется реакция Кумбса, которая производится в двух вариантах
— прямом и непрямом. Для выявления антител, фиксированных на поверхности эритроцитов, производится прямая реакция Кумбса, сущность которой заключается в том, что к сыворотке кролика, иммунизированного глобулинами человеческой сыворотки, добавляется суспензия отмытых в изотоническом растворе хлорида натрия эритроцитов обследуемого: при наличии на поверхности эритроцитов антиэритроцитарных антител происходит агглютинация эритроцитов. Эритроциты лиц, не страдающих приобретенной гемолитической анемией, не агглютинируются. Для выявления конглютининов в начале к сыворотке больного добавляют эритроциты здорового человека, чтобы фиксировать на них антитела, затем их отмывают и проводят с ними прямую реакцию Кумбса. В данном варианте она носит название непрямой.
Течение и осложнения. В большинстве случаев течение волнообразное; обострения наступают под влиянием инфекций, приема больших доз некоторых лекарственных препаратов, например салицилатов, и других преходящих факторов. В тяжелых, длительно протекающих случаях может наступить истощение костного мозга, и анемия принимает гипорегенераторный характер. В ряде случаев подавление активности костного мозга, возможно, обусловлено также выработкой аутоантител к клеткам эритробластического ростка. Осложнением болезни является образование пигментных камней в желчном пузыре. Могут наблюдаться тромбофлебиты, тромбоз селезеночной вены.
Лечение. Проводят терапию кортикостероидами, которые подавляют выработку антиэритроцитарных аутоантител. Переливания крови производят лишь в крайних случаях, так как гемотрансфузия может резко усилить гемолиз.
Миелоапластический синдром объединяет большую группу состояний, различных по этиологии и патогенезу, основные клинические проявления которых обусловлены подавлениемкроветворения. По происхождению различают врожденные, т. е. генетически обусловленные, и приобретенные формы миелоидной аплазии; по течению — острые и хронические. Выделяют также формы, характеризующиеся неполным угнетением регенераторной способности костного мозга, его гипоплазией и полным подавлением его функции— аплазией.
Различные варианты миелоидной гипо- и аплазии, обусловленные преимущественно парциальным (т. е. частичным, в одном направлении) или тотальным (во всех направлениях) подавлением регенераторной способности костного мозга, в клинике выделяются под разными названиями. Наиболее яркими формами являются гипо- и апластическая анемии, при которых в первую очередь подавляется эритропоэтическая функция костного мозга, агранулоцитоз, характеризующийся подавлением гранулоцитопоэтической функции костного мозга, а также панмиелофтиз, при котором нарушена регенеративная функция костномозговой ткани во всех направлениях (т. е. более или менее равномерно нарушена продукция как эритроцитов, так и гранулоцитов и тромбоцитов).
С гипо- и апластическими состояниями костного мозга, в частности с гипопластической анемией, не следует смешивать его гипорегенераторные состояния, развивающиеся, например, при хронической постгеморрагической анемии, тяжелой гемолитической анемии и в некоторых других случаях. В отличие от гипопластических анемий при гипорегенераторных анемиях в костном мозге сохраняется довольно большое количество родоначальных клеток эритробластического ростка, не наблюдается неуклонно прогрессирующего течения болезни: при ликвидации причины, приведшей к
истощению костного мозга, его функция восстанавливается. К миелоапластическим состояниям также не относятся случаи, когда уменьшение содержания в периферической крови форменных элементов (эритроцитов, нейтрофилов, тромбоцитов) обусловлено их повышенным разрушением в селезенке, «гиперспленизмом». Такие состояния наблюдаются при значительном увеличении селезенки, например при циррозах печени, поскольку при этих состояниях активность костного мозга не снижена, а, наоборот, повышена. Не попадают в эту группу и цитопении, обусловленные метаплазией костного мозга (при злокачественных лимфомах, лейкозах) и вытеснением миелопролиферативной ткани (при миеломной болезни, множественных метастазах рака и др.).
Этиология и патогенез. Этиологические факторы миелоидной гипо- и аплазии разнообразны. В одних случаях это эндогенные причины, например гипофункция вилочковой железы, наследственная предрасположенность и др. Однако чаще всего на первом месте находятся экзогенные факторы, среди которых наибольшее значение имеют следующие: а) интоксикация химическими веществами, например отравление бензолом, тетраэтилсвинцом; б) длительный бесконтрольный прием некоторых лекарственных препаратов, например амидопирина, бутадиона, цитостатических средств (эмбихин, ТиоТЭФ, меркаптопурин), метилтиоурацила, сульфаниламидов, некоторых антибиотиков (левомицетин) или повышенная чувствительность к ним; в)факторы (туберкулез, сепсис, сифилис); г) употребление в пищу продуктов растительного происхождения, в которых при определенных условиях могут образоваться токсичные вещества (зерно перезимовавших в поле злаков); д) действие ионизирующей радиации (радиоактивные вещества, рентгеновское излучение и др.). Считается, что действие перечисленных факторов в основном заключается в угнетении ферментов нуклеопротеидного ряда, что приводит к задержке митотического деления клеток.
В последнее время в развитии ряда форм миелоидной гипоплазии и аплазии придается значение аутоагрессивному механизму — выработке организмом антител против собственных клеток крови и костного мозга. Возможно, однако, что выработка аутоантител не является пусковым механизмом болезни, а происходит вторично, к уже измененным клеткам. В ряде случаев причины миелоидной аплазии остаются неясными.
Патологоанатомическая картина. В классических случаях обнаруживаются резкое малокровие органов, дистрофические изменения в них, а также следы множественных кровоизлияний. «Территория» красного костного мозга резко сужена за счет замещения его жировой тканью.
Клиническая картина. В зависимости от преимущественной направленности подавления функции костного мозга клиническая картина различна. Однако наиболее характерные симптомы бывают обусловлены анемизацией, геморрагическим диатезом, появлением некрозов тканей и присоединением вторичной инфекции (например, при агранулоцитозе). В процессе развития заболевания нередко отмечается определенная динамичность клинической картины: если вначале доминировалсиндром, то затем присоединяются другие синдромы и проявления болезни все более свидетельствуют о тотальном подавлении регенераторной способности костного мозга. Если заболевание начинается постепенно и незаметно, первыми обычно являются жалобы на слабость, одышку, немотивированную быструю утомляемость, снижение трудоспособности. При тромбоцитопении возможны геморрагические явления — кровотечения из носа, множественные «синяки» на коже, возникающие при малейшей травме, а иногда и спонтанно,маточные кровотечения и др. Как правило, наблюдается лихорадочное состояние.
При общем осмотре обращает на себя внимание бледность больных; могут быть следы подкожных кровоизлияний в видепятен, в дальнейшем приобретающих бурую, а затем желтую окраску. Кожа влажная, ее тургор нерезко снижен. При выраженной тромбоцитопении бывают положительные симптомы «щипка» и «жгута». Изменения в сердце, легких, почках итракте в
большинстве случаев малохарактерны, однако при возникновении геморрагического синдрома возможны кровоизлияния в различные органы и кровотечения, как внутренние, так и наружные. Лимфатические узлы, печень и селезенка обычно не увеличены.
Картина крови отражает различную направленность и степень нарушения регенераторной способности костного мозга. При гипо- и апластической анемии в крови резко уменьшено содержание эритроцитов — дои менее. Как правило, эритропении сопутствуют более или менее выраженные лейко- и тромбоцитопения. Резко уменьшено количество ретикулоцитов.
При агранулоцитозе наиболее выражена нейтропения: количество лейкоцитов уменьшается до 1,5*10 9 и даже допричем в первую очередь снижается содержание гранулоцитов, которое не превышаетот общего количества лейкоцитов. За счет этого возникают относительный лимфоцитоз и моноцитоз, хотя абсолютное содержание в крови этих клеточных форм лейкоцитов обычно не изменено или незначительно уменьшено. Более молодые формы нейтрофильных лейкоцитов — палочкоядерные — в периферической крови практически отсутствуют. Не удается обнаружить и эозинофилы. Наблюдается ряд патологических изменений в ядрах и цитоплазме нейтрофилов: пикноз ядер, токсическая зернистость цитоплазмы. Анемия и тромбоцитопения в начальном периоде нерезко выражены, однако затем прогрессируют.
Наконец, в некоторых случаях анемия, лейкопения и тромбоцитопения развиваются почти параллельно, что соответствует клинической картине панмиелофтиза (panmyelophthisis — от греч.— pan — «охватывающий все», myelos — костный мозг, При уровне тромбоцитов ниже 9 /л крови возникает геморрагический синдром.
В начальных стадиях заболевания картина стернального пунктата существенно не изменена; может наблюдаться относительное уменьшение клеток эритробластического или миелобластического ростка. В выраженных случаях отмечается бедность стернального пунктата клеточными элементами, преобладают ретикулярные и плазматические клетки. Поскольку аплазия костномозговой ткани развивается неравномерно, в стернальный пунктат могут случайно попасть участки костного мозга с наименее выраженными изменениями, что иногда вводит в заблуждение врача относительно тяжести процесса. Более четкую и точную картину костномозгового кроветворения дает трепанобиопсия гребешка подвздошной кости, позволяющая приготовить гистологические препараты костного мозга.
Течение. Различают острые формы, в ряде случаев с молниеносным течением (описаны случаи агранулоцитоза, когда смерть больных наступала через 2 дня), подострые и хронические формы. Формы с подавлением лейкопоэза в большинстве случаев протекают быстрее, чем с преимущественным подавлением эритропоэза, что в известной степени определяется неравной продолжительностью жизни белых и красных кровяных клеток.
Прогноз крайне неблагоприятен при врожденной и генуинной формах миелоидной аплазии: заболевание прогрессирует и через определенный срок заканчивается смертью больного. При миелотоксической форме болезни, например при передозировке анальгина, бутадиона или цитостатических препаратов, если вовремя прекратить их прием и провести соответствующее лечение, нередко удается приостановить развитие заболевания и даже добиться излечения. При агранулоцитозе частой причиной смерти больных является сепсис.
Лечение. Проводится обязательно в стационарных условиях. С целью воздействия на аутоиммунный механизм развития болезни назначают кортикостероидные препараты, например преднизолон.
При значительной анемизации проводят повторные переливания крови и эритроцитной массы. Больным лейкопенией и тромбоцитопенией переливают не только кровь, но и специально приготовленные лейкоцитную и тромбоцитную массы. Для стимуляции лейкопоэза назначают натрия нуклеинат и пентоксил, анаболические стероидные
источник
— это анемия, развивающаяся в результате усиленного разрушения эритроцитов (преобладание кроверазрушения над кровообразованием).
Наследственные гемолитические анемии
- Мембранопатии (связанные с нарушением мембраны эритроцитов):
— овалоцитоз
— стоматоцитоз
— носительство аномального гемоглобина
Приобретенные гемолитические анемии
Могут быть связанны с воздействием АТ к эритроцитам и эритроидным клеткам КМ; повреждением оболочки эритроцитов вследствие протезирования клапанов сердца; хроническое повреждение эритроцитов ядами, тяжёлыми металлами; недостаток витамина Е и др. причины.
Внутриклеточный гемолиз – разрушение в клетках фагоцитарной системы.
Внутрисосудистый гемолиз – в сосудистом русле с участием комплимента.
| Внутриклеточный гемолиз | Внутрисосудистый гемолиз | |
| Локализация гемолиза | В макрофагах селезенки | В сосудах |
| Размеры селезенки | Увеличена, безболезненна | В остром периоде может быть увеличена |
| Изменения эритроцитов | Характерно | — |
| Высокий уровень свободного гемоглобина в крови | — | Характерно |
| Гемоглобинурия и гемосидеринурия | — | Характерно |
- Гиперрегенераторный тип эритропоэза, что проявляется увеличением количества эритроидных клеток в КМ и повышением количества ретикулоцитов в периферической крови.
- Повышение уровня свободного билирубина в крови, проявляется желтухой.
- Гиперхолия кала и наличие уробилина в моче
При данной анемии, сущность патологического процесса заключается в отсутствие белка спектрина в мембране эритроцитов. При этом эритроциты будут меньшего диаметра и имеют не двояковогнутую форму, а форму двояковыпуклой линзы. Мембрана таких эритроцитов имеет повышенную проницаемость для ионов натрия, это приводит к их набуханию.
Продолжительность жизни таких эритроцитов- 8-15 дней.
Первые признаки заболевания проявляются в юношеском возрасте.
Течение волнообразное, провоцирующими факторами являются инфекции, переохлаждение, беременность.
— Желтушный синдром. В течение длительного времени единственным признаком является желтушное окрашивание кожи и склер. Со временем усиливается.
Изменение костей черепа, высокое «готическое» небо, зубные аномалии, полидактилия, синдактилия, микрофтальмия.
При частых обострениях – спленэктомия.
Это форма гемоглобинопатии, при которой формируется HbS. Установлено, что растворимость HbS при отдаче кислорода уменьшается в 100 раз, что приводит к образованию геля, а эритроциты принимают форму серпа. Деформированные эритроциты ригидны, у них нет нормальной пластичности и они начинают закупоривать мелкие кровеносные сосуды, что создает условия для развития ишемии и некрозов.
Заболевание проявляется через несколько месяцев после рождения. Течение хроническое и характеризуется повторными гемолитическими кризами.
При обследовании таких больных, кроме гематологических изменений, имеется непропорциональный высокий рост, башенный череп, инфантилизм, трофические язвы голеней, ЖКБ, фиброзные изменения в печени, селезенке, миокарде.
Большинство больных умирают в детстве.
Лечение носит симптоматический характер. Кроме обезболивающих, применяют адекватное обеспечение больного жидкостью, установлено, что гемодилюция уменьшает возможность образования серповидных эритроцитов.
В норме синтез цепей глобина сбалансирован. Нарушение синтеза одной из цепи приводит к нарушению баланса. Цепь, которая продуцируется избыточно, откладывается в эритроцитах , что ведет к повреждению мембраны эритроцитов и ускоренному их разрушению. Различают α и β талассемию.
— Анемический синдром (гипохромная анемия)
— Среди эритроцитов преобладают микросфероциты с повышенной оматической резистентностью.
Дети отстают в физическом развитии. Аномалии в развитии скелета: монголойдные черты лица, башенный череп.
Продолжительность жизни 5-8 лет.
Главной опасностью для больных талассемией является инфекция и малокровие. Дети нуждаются в полноценном питании, назначении фолиевой кислоты.
источник
Учебно-методическое пособие для студентов по теме:
«Гемолитические анемии. Агранулоцитоз»
Гемолитические анемии представляют собой обширную группу заболеваний, различающихся по этиологии, патогенезу, клинической картине, методам лечения. Основным признаком гемолитических анемий является повышенный распад эритроцитов и укорочение продолжительности их жизни. В физиологических условиях продолжительность жизни эритроцитов составляет 100-120 дней. Стареющие эритроциты подвергаются секвестрации в синусах селезенки, а также в костном мозге. Образовавшийся в результате физиологического распада эритроцитов пигмент билирубин циркулирует в крови в виде свободного (неконъюгированного) билирубина, который транспортируется в печеночную клетку, где при участии ферментов соединяется с глюкуроновой кислотой. Образовавшийся билирубин-глюкуронид (конъюгированный) проникает из печеночных клеток в желчные ходы и выделяется вместе с желчью в кишечник. При гемолитических анемиях вследствие усиленного разрушения эритроцитов продолжительность их жизни укорачивается до 12 – 14 дней. Патологический гемолиз может быть преимущественно внутрисосудистым. Внутриклеточный распад эритроцитов происходит в клетках ретикулогистиоцитарной системы, главным образом в селезенке, и сопровождается повышением в сыворотке свободного билирубина, увеличением экскреции уробилина с мочой и калом, наклонностью к образованию камней в желчном пузыре и протоках. При внутрисосудистом гемолизе гемоглобин поступает в повышенном количестве в плазму и выделяется с мочой в неизмененном виде или в виде гемосидерина, который может откладываться во внутренних органах (гемосидероз). По течению гемолиз может быть острым или хроническим. Все гемолитические анемии делятся на две большие группы: наследственные и приобретенные.
Наследственные гемолитические анемии являются следствием различных генетических дефектов в эритроцитах, которые становятся функционально неполноценными и нестойкими.
Приобретенные гемолитические анемии связаны с воздействием различных факторов, способствующих разрушению эритроцитов (образование антител, гемолитические яды, механические воздействия и прочее).
Наледственный микросфероцитоз (болезнь Минковского – Шоффара)
Наследственный микросфероцитоз был впервые описан в 1900 году Минковским, а в дальнейшем более подробно – Шоффаром.
В основе заболевания лежит генетический дефект белка мембраны эритроцита. Имеющаяся аномалия мембраны приводит к проникновению в эритроцит избытка ионов натрия и повышенному накоплению в нем воды, вследствие чего образуются сферические эритроциты (сфероциты). Сфероциты, в отличие от двояковогнутых нормальных эритроцитов, не обладают способностью деформироваться в узких участках кровотока, например при переходе в синусы селезенки. Это ведет к замедлению продвижения эритроцитов в синусах селезенки, отщеплению части поверхности эритроцита с образованием микросфероцитов (отсюда название болезни – микросфероцитоз) и постепенной их гибели. Разрушенные эритроциты поглощаются макрофагами селезенки. Постоянный гемолиз эритроцитов в селезенке ведет к гиперплазии клеток ее пульпы и увеличению органа. В связи с усиленным распадом эритроцитов в сыворотке повышается содержание свободного билирубина. Поступающий в повышенном количестве в кишечник билирубин выводится из организма с мочой и главным образом с калом в виде стеркобилина. Суточное выделение стеркобилина при наследственном микросфероцитозе превышает норму в 10-20 раз. Следствием повышенного выделения билирубина в желчь является плейохромин желчи и образование пигментных камней в желчном пузыре и протоках.
Кожа и внутренние органы при наследственном микросфероцитозе бледны и желтушны. Костный мозг в плоских и трубчатых костях гиперплазирован за счет эритроидного ростка, отмечаются явления эритрофагоцитоза ретикулярными клетками. В селезенке наблюдаются резко выраженное кровенаполнение пульпы, гиперплазия эндотелия синусов, уменьшение размеров и количества фолликулов. В печени, костном мозге, лимфатических узлах нередко выявляется гемосидероз.
Клиника зависит от выраженности гемолиза. В большинстве случаев первые признаки выявляются в юношеском или зрелом возрасте. У детей болезнь обнаруживается обычно при обследовании по поводу заболевания их родственников. Жалобы вне обострения заболевания могут отсутствовать. В период обострения отмечаются слабость, головокружение, повышение температуры. Одним из основных клинических симптомов является желтуха, которая долгое время может оставаться единственным признаком заболевания. Выраженность желтухи зависит, с одной стороны, от интенсивности гемолиза, а с другой – от способности печени к конъюгированию свободного билирубина с глюкуроновой кислотой. В моче билирубин не обнаруживается, так как свободный билирубин не проходит через почки. Кал интенсивно окрашен в темно-коричневый цвет вследствие повышенного содержания стеркобилина. В связи со склонностью к камнеобразованию у больных могут наблюдаться приступы желчнокаменной болезни, нередко сопровождающиеся признаками холецистита. В случае закупорки камнем общего желчного протока возникает синдром обтурационной желтухи (значительное повышение содержания билирубина наличие желчных пигментов в моче, кожный зуд и так далее). Кардинальным симптомом наследственого микросфероцитоза является увеличение селезенки, которая обычно выступает из-под подреберья на 2–3 см. При длительно протекающем гемолизе наблюдается значительная спленомегалия, в связи с чем больные жалуются на тяжесть в левом подреберье. Печень при неосложненном заболевании обычно нормальных размеров, но иногда у больных, длительно страдающих гемолитической анемией, обнаруживается ее увеличение. Могут наблюдаться признаки замедленного развития, а также нарушения лицевого скелета в виде «башенного черепа», седловидного носа, высокого стояния неба, нарушения расположения зубов, узких глазниц. Выраженность анемического синдрома различна. Часто отмечается умеренное снижение гемоглобина. У некоторых больных анемия вообще отсутствует. Наиболее резкая анемизация наблюдается в период гемолитических кризов. У лиц среднего и пожилого возраста иногда встречаются плохо поддающиеся лечению трофические язвы голени, связанные с агглютинацией и распадом эритроцитов в мелких капиллярах конечностей. Течение заболевания характеризуется так называемыми гемолитическими кризами, проявляющимися резким усилением симптомов на фоне непрерывно текущего гемолиза. При этом повышается температура в связи с массовым распадом эритроцитов, увеличивается интенсивность желтухи, появляется сильные боли в животе, рвота. Гемолитические кризы возникают обычно после интеркуррентных инфекций, переохлаждения, у женщин в связи с беременностью. Частота кризов различна, у ряда больных они не возникают.
Анемия при наследственном микросфероцитозе носит нормохромный характер. В мазке крови преобладают микросфероциты, отличающиеся отсутствием характерного для нормальных эритроцитов центрального просветления. Преобладание микроцитов выявляется графически на кривой Прайс-Джонса, отражающей количественные соотношения эритроцитов различных диаметров (средний диаметр нормального эритроцита составляет 7–7,5 мкм). При наследственном микросфероцитозе вершина кривой Прайс-Джонса растянута и сдвинута влево в сторону микроцитов. Количество ретикулоцитов увеличено. Число лейкоцитов обычно нормально. При гемолитических кризах отмечается нейтрофильный лейкоцитоз со сдвигом влево. Количество тромбоцитов варьирует в пределах нормы. В костном мозге отмечается выраженная гиперплазия эритроидного ростка. Содержание непрямого билирубина в крови повышено умеренно и, как правило, не превышает 50–70 мкмоль/л. Определяется повышенное содержание уробилина в моче и стеркобилина в кале. Диагноз наследственного микросфероцитоза ставится на основании течения заболевания (чередование кризов и ремиссий), клинической картины (желтуха, спленомегалия, боли в правом подреберье, анемия), данных исследования периферической крови (нормохромная анемия, ретикулоцитоз, микросфероцитоз). Важное значение имеет обследование родственников больных, у которых могут определяться едва уловимые признаки гемолиза или микросфероцитоз без клинических проявлений. Дополнительными диагностическими критериями может служить ряд лабораторных тестов. Характерным лабораторным признаком заболевания является снижение осмотической резистентности эритроцитов по отношению к гипотоническим растворам хлористого натрия. Начало гемолиза при наследственном микросфероцитозе соответствует 0,6–0,7%, а конец – 0,4% вместо 0,48 и 0,22% в норме. Снижение осмотической резистентности свидетельствует о преобладании в крови эритроцитов сферической формы – сфероцитов, которые, менее стойки к осмотическому гемолизу, чем нормальные макропланоциты. Правильному диагнозу способствует проба Кумбса, выявляющая фиксированные на эритроцитах аутоантитела при аутоиммунных гемолитических анемиях.
Единственным методом лечения больных наследственным микросфероцитозом является спленэктомия, которая оказывается эффективной в 100% случаях. После спленэктомии у больных наступает практическое излечение, несмотря на то, что эритроциты сохраняют свои патологические свойства (микросфероцитоз, снижение осмотической резистентности). Прекращение гемолиза после спленэктомии объясняется удалением основного плацдарма разрушения микросфероцитов. Спленэктомия показана при частых гемолитических кризах, резкой анемизации больных, инфарктах селезенки, приступах печеночной колики. При наличии соответствующих показаний в некоторых случаях одновременно со спленэктомией может быть произведена холецистэктомия. При легких компенсированных формах заболевания у взрослых показания к спленэктомии следует ограничивать. В качестве предоперационной подготовки анемизированных больных показаны переливания эритроцитарной массы. Глюкокортикоидные гормоны при наследственном микросфероцитозе неэффективны. Прогноз при наследственном микросфероцитозе относительно благоприятен. Многие больные доживают до старости.
Наследственные гемолитические анемии, связанные с дефицитом активности ферментов
Эта неоднородная группа заболеваний обозначается также как несфероцитарные гемолитические анемии. В отличие от микросфероцитоза они характеризуются нормальной формой эритроцитов с тенденцией к макропланоцитозу, нормальной или повышенной осмотической резистентностью эритроцитов, рецессивным типом наследования, отсутствием эффекта от спленэктомии.
В основе патогенеза несфероцитарных гемолитических анемий лежит дефицит активности некоторых ферментов эритроцитов, в результате чего эритроциты становятся чувствительными к воздействию различных веществ растительного происхождения, лекарственных средств. Наиболее распространенной среди этой группы заболеваний является острая гемолитическая анемия, связанная с дефицитом глюкозо‑6‑фосфатдегидрогеназы (Г‑6‑ФДГ). Согласно сведениям ВОЗ, в мире насчитывается около 100 миллионов человек с дефицитом активности Г‑6‑ФДГ. Дефицит Г‑6‑ФДГ наследуется по рецессивному типу, сцепленному с полом, в связи с чем клинические проявления данной патологии наблюдаются преимущественно у мужчин. При низкой активности Г‑6‑ФДГ в эритроцитах нарушаются процессы восстановления никотинамиддинуклеотидфосфата (НАДФ) и превращения окисленного глютатиона в восстановленный, предохраняющий эритроцит от разрушающего действия потенциальных гемолитических агентов (фенилгидразин, некоторые медикаменты, бобовые и т.д.). Гемолиз происходит преимущественно внутрисосудисто. Кожа и внутренние органы желтушны. Отмечается увеличение и полнокровие печени и селезенки, умеренное увеличение и набухание почек. Микроскопически в почечных канальцах обнаруживают гемоглобинсодержащие цилиндры. В печени и селезенке наблюдается макрофагальная реакция с наличием в макрофагах гемосидерина.
Как правило, дефицит Г‑6‑ФДГ не проявляется клинически без воздействия различных гемолитических агентов. Спровоцировать гемолитический криз могут противомалярийные препараты, сульфаниламиды, анальгетики, некоторые химиопрепараты (фурадонин, ПАСК), витамин К, растительные продукты (бобовые, стручковые). Выраженность гемолитического процесса зависит от степени дефицита Г‑6‑ФДГ и от дозы принятого препарата. Гемолиз наступает не сразу, а через 2–3 дня после приема препаратов. В тяжелых случаях у больных появляется высокая температура резкая слабость, боли в животе и спине, обильная рвота. Отмечается выраженная одышка, сердцебиение, нередко развитие коллаптоидного состояния. Характерным симптомом является выделение темной мочи, имеющей иногда черный цвет, что связанно с внутрисосудистым распадом эритроцитов и выделением с мочой гемосидерина. В некоторых случаях вследствие закупорки почечных канальцев продуктами распада гемоглобина и резкого снижения клубочковой фильтрации возможно развитие острой почечной недостаточности. При объективном исследовании отмечается желтушная окраска кожных покровов и слизистых оболочек, увеличение селезенки, реже печени. Через неделю гемолиз прекращается, независимо от того, продолжается прием препарата или нет.
В течение первых двух суток гемолитического криза у больных развивается выраженная нормохромная анемия с падением гемоглобина до 30 г./л и ниже. Отмечается высокий ретикулоцитоз, наличие нормоцитов в крови. Особенностью эритроцитов является присутствие в них телец Гейнца, представляющих собой денатурированный гемоглобин и выявляющихся при суправитальной окраске. Осмотическая резистентность эритроцитов нормальная или повышена. Со стороны белой крови во время криза отмечается лейкоцитоз со сдвигом влево до миелоцитов и более молодых форм. В костном мозге наблюдается гиперплазия эритроидного ростка и явления эритрофагоцитоза. Диагноз острой гемолитической анемии, связанной с дефицитом Г‑6‑ФДГ, ставится на основании типичной клинико-гематологической картины острого внутрисосудистого гемолиза, связи заболевания с приемом лекарств и данных лабораторных исследований, выявляющих снижение активности Г‑6‑ФДГ в эритроцитах больных, а иногда их родственников. Основным методом лечения острой гемолитической анемии при выраженном падении содержания гемоглобина являются повторные переливания свежецитратной одногруппной крови по 250–500 мл 1–2 раза в неделю внутривенные вливания больших количеств физиологического раствора или 5% раствора глюкозы. В качестве противошоковых препаратов применяют морфин, преднизолон, промедол. Из сосудистых средств используют кордиамин, камфору. При развитии острой почечной недостаточности проводят обычный комплекс терапевтических мероприятий, при отсутствии эффекта показано проведение гемодиализа. При нетяжелых гемолитических кризах в качестве антиоксидантного препарата назначают эревит внутримышечно по 2 мл 2 раза в день. Профилактика гемолитических кризов заключается в тщательном сборе анамнеза перед назначением средств, способных спровоцировать гемолитический криз при дефиците Г‑6‑ФДГ. При необходимости применения этих препаратов у лиц с дефицитом Г‑6‑ФДГ рекомендуется использовать средства для восстановления глютатиона. С этой целью применяют ксилит в суточной дозе 30 г. в комбинации с рибофлавином в дозе 0,03 г. в течение 1–2 месяцев. Прогноз неблагоприятен при развитии анурии и почечной недостаточности. При молниеносных формах заболевания смерть наступает от шока или острой аноксии.
Аутоиммунная гемолитическая анемия является наиболее часто встречающимся заболеванием среди приобретенных гемолитических анемий. Развитие заболевания связано с появлением в организме больного антител к собственным эритроцитам, которые агглютинируются и подвергаются распаду в клетках ретикулогистиоцитарной системы.
Различают симптоматические и идиопатические аутоиммунные гемолитические анемии. Симптоматические аутоиммунные анемии возникают на фоне различных заболеваний, сопровождающихся нарушениями в иммунокомпетентной системе. Наиболее часто они встречаются при хроническом лимфолейкозе, лимфогранулематозе, остром лейкозе, системной красной волчанке, ревматоидном артрите, хронических гепатитах и циррозах печени. В тех случаях, когда появление аутоантител не удается связать с каким-либо патологическим процессом, говорят об идиопатической аутоиммунной гемолитической анемии, которая составляет около 50% всех аутоиммунных анемий. Образование аутоантител происходит в результате нарушения в системе иммунокомпетентных клеток, которые воспринимают эритроцитарный антиген как чужеродный и начинают вырабатывать к нему антитела. После фиксации аутоантител на эритроцитах последние захватываются клетками ретикулогистиоцитарной системы, где подвергаются агглютинации и распаду. Гемолиз эритроциов происходит главным образом в селезенке, печени, костном мозге. Аутоантитела к эритроцитам принадлежат к различным типам. По серологическому принципу аутоиммунные гемолитические анемии делятся на несколько форм:
· анемии с неполными тепловыми агглютининами;
· анемии с тепловыми гемолизинами;
· анемии с полными холодовыми агглютининами;
· анемии с двухфазными гемолизинами;
· анемии с агглютининами против нормобластов костного мозга.
Каждая из этих форм имеет некоторые особенности в клинической картине, течении и серологической диагностике. Наиболее часто встречаются анемии с неполными тепловыми агглютининами, составляющие 70–80% всех аутоиммунных гемолитических анемий.
По клиническому течению выделяют острую и хроническую аутоиммунную гемолитическую анемию. При острых формах у больных внезапно появляется резкая слабость, сердцебиение, одышка, лихорадка, желтуха. При хронических формах заболевание развивается исподволь. Общее состояние больных изменяется мало. Одышка и сердцебиение могут отсутствовать, несмотря на выраженную анемизацию, что связано с постепенной адаптацией больных к гипоксии. Объективно выявляется увеличение селезенки, реже – печени. При аутоиммунной анемии, связанной с холодовыми агглютининами (холодовая агглютининовая болезнь), отмечается плохая переносимость холода и развитие на холоду таких симптомов, как крапивница, синдром Рейно, гемоглобинурия. Течение заболевания характеризуется наклонностью к обострениям (гемолитическим кризам) под влиянием инфекций чаще вирусных или при воздействии холода. Отмечается нормохромная или умеренно гиперхромная анемия различной степени, ретикулоцитоз, нормоцитоз. Для аутоиммунной гемолитической анемии с холодовыми агглютининами характерна агглютинация эритроцитов, наблюдающаяся сразу после взятия крови и в мазке. При подогревании агглютинация исчезает. Осмотическая резистентность эритроцитов в большинстве случаев снижена. Количество лейкоцитов при идиопатических формах подвержено колебаниям в случае острого гемолитического криза встречается лейкоцитоз со сдвигом влево до миелоцитов. При хронических формах количество лейкоцитов, близко к норме. Число тромбоцитов не изменено. СОЭ сильно увеличена. В костном мозге наблюдается резко выраженная гиперплазия эритроидного ростка. Среди лабораторных признаков повышенного гемолиэа отмечается увеличение содержания непрямого билирубина, повышенная экскреция стеркобилина с калом.
Диагноз аутоиммунной гемолитической анемии ставится на основании признаков повышенного гемолиза, с одной стороны, и выявления фиксированных на поверхности эритроцита антител – с другой. Основным методом выявления на эритроцитах аутоантител является проба Кумбса, основанная на преципитации антиглобулиновой сыворотки эритроцитов с фиксированными на них антителами. Различают прямую и непрямую пробы Кумбса. Прямая проба выпадает положительной в большинстве случаев аутоиммунной гемолитической анемии. Отрицательный результат прямой пробы означает отсутствие антител на поверхности эритроцита и не исключает наличия свободных циркулирующих антител в плазме. Для выявления свободных антител применяют непрямую пробу Кумбса.
Средством выбора при лечении аутоиммунной гемолитической анемии являются глюкокортикоидные гормоны, которые практически всегда прекращают или уменьшают гемолиз. Необходимым условием гормональной терапии является достаточная дозировка и длительность. В острой фазе назначают преднизолон в больших дозах – 60–80 мг/сут (из расчета 1 мг/кг массы) или эквивалентные дозы других глюкокортикоидов. После наступления ремиссии доза преднизолона постепенно уменьшается. Поддерживающая доза составляет 5-10 мг/сут. Лечение проводится на протяжении 2-3 месяцев, до исчезновения всех признаков гемолиза и негативации пробы Кумбса. У некоторых больных эффект оказывают иммунодепрессанты (6‑меркаптопурин, азатиоприн, хлорамбуцил), а также противомалярийные препараты (делагил, резохин). Особенно выраженный эффект иммунодепрессанты оказывают при аутоиммунной гемолитической анемии, связанной с холодовыми агглютининами. Доза 6‑меркаптопурина и азатиоприна составляет 100–150 мг/сут, хлорамбуцил назначают в дозе 10–15 мг. При рецидивирующих формах заболевания и отсутствии эффекта от применения глюкокортикоидов и иммунодепрессантов показана спленэктомия. Гемотрансфузии у больных аутоиммунной гемолитической анемией следует проводить только по жизненным показаниям (резкое падение гемоглобина, сопорозное состояние). Рекомендуется специально подбирать доноров, чьи эритроциты дают отрицательную пробу Кумбса.
Геомолитическая анемия с постоянной гемосидеринурией и пароксизмальной ночной гемоглобинурией (болезнь Маркиафавы – Микели)
Представляет собой приобретенную гемолитическую анемию с постоянным внутрисосудистым гемолизом и выделением с мочой гемосидерина.
Заболевание рассматривается как результат соматической мутации эритроидных клеток, вследствие чего вырабатывается патологический клон (семейство) эритроцитов с повышенной чувствительностью к различным гемолитическим агентам. Интенсивность гемолиза повышается под влиянием пропердина, комплемента, тромбина, ацидоза.
Характерным морфологическим признаком заболевания является гемосидероз почек. Гемосидероза в других органах не наблюдается. В печени отмечаются дистрофические и некробиотические изменения, связанные с венозными тромбозами и анемией. Селезенка увеличена, за счет множественных сосудистых тромбозов и развития периваскулярного склероза.
Жалобы больных сводятся к слабости, головокружениям, одышке, сердцебиениям. Характерным признаком являются боли в животе различной локализации и интенсивности, наблюдающиеся, как правило, в период криза и связанные с капиллярными тромбозами мезентериальных сосудов. Нередко отмечаются тромбозы периферических сосудов, чаще в венах верхних и нижних конечностей, а также других сосудов (мозговых, селезеночных, почечных). В период криза у больных может повышатся температура. Типичным признаком заболевания является появление мочи черного цвета, обусловленного выделением с мочой гемосидерина и гемоглобина. Нередко гемоглобинурия имеет место в ночное время (пароксизмальная ночная гемоглобинурия). Этот феномен объясняется наступающим во время сна физиологическим ацидозом, активацией пропердина и других факторов, усиливающих гемолиз. Гемоглобинурия не является обязательным симптомом заболевания. При объективном исследовании выявляется бледность кожи с небольшим желтушным оттенком, умеренное увеличение селезенки и печени.
Анемия в течение длительного времени носит нормохромный характер. При значительных потерях с мочой железа в виде гемоглобина и гемосидерина цветовой показатель становится меньше нормы. Количество ретикулоцитов повышено незначительно. Часто наблюдается лейкопения и тромбоцитопения. В костном мозге отмечается гиперплазия эритроидного ростка, сопровождающаяся нередко угнетением гранулоцитарного и мегакариоцитарного ростков. О возможности болезни Маркиафавы – Микели следует думать у больных с гемолитической анемией, резистентной к лечению, сопровождающейся лейко- и тромбоцитопенией без значительного увеличения селезенки. Важное диагностическое значение имеет микроскопирование осадка мочи с целью выявления гемосидеринурии. Среди лабораторных тестов в диагностике болезни Маркиафавы-Микели используются следующие тесты: кислотный тест Хэма (гемолиз эритроцитов больного в подкисленной человеческой сыворотке), тест Кросби (усиление гемолиза под влиянием тромбина), сахарозный тест (гемолиз эритроцитов больных в свежей донорской крови при добавлении сахарозы).
Наиболее эффективным методом лечения является переливание эритроцитов, предварительно трижды отмытых физиологическим раствором хлорида натрия. Такие эритроциты переливают раз в 4–5 дней, в количестве 200–400 мл, причем они должны быть перелиты не позднее 48 ч после взятия. Для профилактики и лечения тромбозов показана антикоагулянтная терапия. В период гемолитического криза некоторое уменьшение гемолиза достигается при введении плазмозаменителей, в частности декстрана или полиглюкина в количестве 500–1000 мл. Глюкокортикоиды и препараты железа при болезни Маркиафавы – Микели противопоказаны.
Агранулоцитоз – синдром, при котором происходит полное или почти полное исчезновение из периферической крови гранулоцитов (зернистых лейкоцитов). Чаще всего агранулоцитоз встречается у взрослых, но он может быть и в детском возрасте.
Различают агранулоцитоз миелотоксический, то есть возникающий при нарушении образования гранулоцитов в костном мозге, и иммунный – при ускоренной гибели гранулоцитов под действием антилейкоцитарных антител.
Нарушение образования гранулоцитов в костном мозге может иметь место при ионизирующем облучении, при лечении противоопухолевыми препаратами, под влиянием продуктов жизнедеятельности грибка типа Fusarium, размножающегося в перезимовавшем зерне.
Антилейкоцитарные антитела при иммунном агранулоцитозе могут образовываться в процессе лечения некоторыми лекарственными препаратами: амидопирином, бутадионом, фенобарбиталом, винбластином, сульфаниламидами, ПАСКом. При иммунном агранулоцитозе стволовые клетки не страдают.
Для любого агранулоцитоза характерны общая слабость, лихорадка, гингивит, стоматит, язвенно-некротические поражения слизистых оболочек полости рта, глотки, желудочно-кишечного тракта. Возможно увеличение печени, селезенки, лимфатических узлов, развитие пневмонии, флегмон.
При миелотоксическом агранулоцитозе, вследствие снижения в крови тромбоцитов, выражен геморрагический синдром (синяки, кровоточивость десен, носовые, желудочные, кишечные кровотечения), поражения слизистых оболочек (некрозы, молочница), полости рта и желудочно-кишечного тракта. В периферической крови уменьшено количество всех форм лейкоцитов, а также тромбоцитов, может быть анемия.
При иммунном агранулоцитозе лейкопения носит умеренный характер, но количество гранулоцитов уменьшено до нуля. Тромбоцитопения отсутствует. В сыворотке крови определяются антилейкоцитарные антитела.
Среди возможных осложнений агранулоцитоза – сепсис, пневмонии, прободение кишечника.
Диагноз агранулоцитоза основывается на клинических проявлениях заболевания, характерной картине периферической крови, результатах исследования пунктата костного мозга.
Прогноз зависит от тяжести процесса и индивидуальных особенностей организма. Если причины агранулоцитоза известны, необходимо их срочно ликвидировать, например отменить токсические препараты. Используют кортикостероиды (при иммунном агранулоцитозе), повторные переливания лейкоцитарной массы, антибиотики, витамины.
При язвенно-некротических поражениях слизистых оболочек полости рта проводят последовательную обработку их раствором перманганата калия (1:5000) и 2%-м раствором перекиси водорода, облепиховым маслом.
1. Внутренние болезни под редакцией Ф.И. Комарова. М.: Медицина. — 1991 г.
2. Маколкин В.И., Овчаренко В.И. Внутренние болезни. М.: Медицина. — 1994 г.
3. Внутренние болезни (в двух томах) под редакцией А.В. Сумарокова. М.: Медицина. — 1993 г.
4. Практические навыки терапевта под редакцией Г.П. Матвейкова. Минск, Вышэйшая школа. — 1993 г.
5. Машковский М.Д. Лекарственные средства (в двух томах). М.: Медицина. — 1993 г.
6. Окороков А.Н. Лечение болезней внутренних органов: Практическое руководство: в 3 т. – Мн.: Выш. шк., Белмедкнига. 1995–1996 гг.
источник