Альфа-талассемия распространена преимущественно в Юго-Восточной Азии, Китае, Африке и в Средиземноморье. Синтез а-цепей кодируют 4 гена, поэтому степень нарушения их синтеза меньше, чем при ß-талассемии; выраженный дисбаланс развивается только тогда, когда поражены все 4 гена. В то же время агрегаты из β-цепей, количество которых при α-талассемии обнаруживают в избытке, более растворимы, чем агрегаты из α-цепей, поэтому гемолиз при α-талассемии выражен слабее, чем при ß-талассемии, а эритропоэз более эффективен. Следовательно, клинические и лабораторные данные при α-талассемии выражены менее отчетливо, чем при ß-талассемии; их основное отличие в биохимическом составе гемоглобина эритроцитов: при α-талассемии уменьшено содержание α-цепей гемоглобина.
Качественные гемоглобинопатии представлены прежде всего серповидно-клеточной анемией, реже — анемией, обусловленной наличием нестабильных гемоглобинов; гомозиготной анемией СС, ЕЕ и др.; бессимптомной гемоглобинопатией НЬС и др., а также обусловленной присутствием гемоглобина М-группы. Более полно изучена серповидноклеточная анемия, широко распространенная у жителей тропической Африки, реже она встречается у жителей Средиземноморья, Ближнего и Среднего Востока, в Южной Америке. Важно отметить, что она распространена преимущественно в регионах, неблагополучных по малярии.
Качественные изменения гемоглобина состоят в том, что в положении ß-цепи глутаминовая кислота заменена на валин. Патологический гемоглобин получил название от слова «sicsle» — серп, потому что эритроцит приобретает серповидную форму. HbS менее растворим, чем нормальный, и в условиях гипоксии выпадает в осадок. В части случаев наблюдается бессимптомное носительство HbS, когда его концентрация в эритроцитах составляет не более 20-45 %; в обычных условиях гемолиз не развивается, спровоцировать его могут лишь особые ситуации, например полет на самолетах или подводное плавание, когда возникает гипоксия. Патогенез гемолиза сложен. Полагают, что в условиях кислой среды в молекуле HbS происходит смена отрицательного заряда на нейтральный, поскольку валин имеет нейтральную реакцию, а глутаминовая кислота — кислую. При этом растворимость гемоглобина снижается, образуются гидрофобные связи между остатками валина, а молекулы патологического HbS кристаллизуются, что приводит к образованию серповидной формы эритроцита.
Часть деформированных эритроцитов изменяется необратимо и преждевременно разрушается фагоцитами. Циркулируя в крови, серповидные эритроциты склеиваются между собой, особенно в мелких сосудах, образуя тромбы и нарушая кровообращение. Возникшая в этих условиях гипоксия способствует образованию новых серповидных эритроцитов — развивается своего рода порочный круг, результатом которого являются стаз и тромбоз в различных органах, в т.ч. в селезенке, которая постепенно атрофируется после гипертрофии. Генетический дефект локализуется в структурном гене ß-цепи, где произошла точечная мутация.
Клиника. Серповидноклеточная анемия клинически характеризуется симптомами, вызванными, с одной стороны, тромбозом сосудов различных органов серповидными эритроцитами, а с другой — гемолитической анемией. Степень тяжести анемии зависит от концентрации HbS в эритроците: чем она больше, тем ярче и тяжелее симптоматика. Кроме того, в эритроцитах могут присутствовать и другие патологические гемоглобины: HbF, HbD, НЬС и др. Иногда серповидноклеточная анемия сочетается с талассемией, при этом клинические проявления могут уменьшаться или, напротив, нарастать.
В первоначальном периоде болезни поражается преимущественно костномозговая система: появляется припухлость, а также боль за счет тромбоза сосудов, питающих сустав и кость. Возможен асептический некроз головки бедренной кости с присоединением в дальнейшем инфекции и остеомиелита. Гемолитические кризы развиваются обычно после перенесенных инфекций, имеют регенераторный либо гипорегенераторный характер и являются основной причиной смерти этих больных. В редких случаях наблюдается секвестрационный криз за счет депонирования крови в селезенке и печени, который выражается болевым абдоминальным синдромом за счет быстрого увеличения этих органов и сопровождается коллапсом; при этом гемолиз может отсутствовать, встречается легочный инфаркты в связи с нарушением микроциркуляции на уровне легочных сосудов.
Во втором периоде постоянный симптом — гемолитической анемии. Развивающаяся в трубчатых костях гиперплазия костного мозга (в них совершается активное кроветворение как компенсаторная реакция на гемолиз) сопровождается характерными изменениями скелета: тонкие конечности, искривленный позвоночник, башенный череп с выпуклостями в области лба и теменной кости. Гепато- и спленомегалия развиваются за счет активации в них эритропоэза, а также вторичного гемохроматоза и тромбоза; у части больных формируется желчнокаменная болезнь. Гемосидероз сердечной мышцы приводит к сердечной недостаточности, а гемосидероз печени, поджелудочной железы — к циррозу печени и сахарному диабету.
Тромбоз сосудов почек протекает с гематурией и последующей почечной недостаточностью. Неврологическая симптоматика обусловлена инсультом, параличом черепных нервов и др. Характерны трофические язвы на нижних конечностях. Большинство больных с тяжелой формой серповидноклеточной анемии погибает в течение 5 лет, а пережившие этот срок вступают в третий период, который характеризуется признаками нерезко выраженной гемолитической анемии. Селезенка у них обычно не прощупывается, так как повторные инфаркты приводят к ее сморщиванию — аутоспленэктомии. Печень остается увеличенной, неравномерно уплотненной, а частые инфекции принимают нередко септическое течение.
Гематологические изменения. Концентрация гемоглобина снижается ( 9 /л. В костном мозге наблюдается гиперплазия эритроидного ростка. Для выявления серповидности эритроцитов проводят специальную пробу: каплю крови покрывают стеклом, герметизируют, для чего края стекла смазывают вазелином; через несколько минут парциальное давление кислорода в капле крови под стеклом снижается и эритроциты принимают серповидную форму. Более информативен электрофорез гемоглобина: при серповидноклеточной анемии у гомозигот основную массу составляет HbS, HbA отсутствует, содержание HbF повышено; у гетерозигот при электрофорезе наряду с HbS выявляют НЬА. В крови повышено содержание свободной фракции билирубина, увеличено содержание сывороточного железа; ОРЭ повышена.
Гетерозиготные больные чувствуют себя практически здоровыми; анемию и морфологические изменения эритроцитов обнаруживают у них только в условиях гипоксии (подъем в горы, тяжелая физическая нагрузка, полет на самолетах и т.п.). Однако гемолитический криз и у них может закончиться летально.
Таким образом, клиника серповидноклеточной анемии характеризуется полисимптомностью: желтушностью кожных покровов, гипоксическим синдромом, гепатоспленомегалией, деформацией скелета, повторным тромбозом органов; из гематологических симптомов: анемией регенераторного характера, серповидностью эритроцитов, выявляемой при специальных пробах, гипербилирубинемией за счет свободной фракции. Принадлежность человека к определенной этнической группе дает основание заподозрить это заболевание и начать целенаправленное обследование для подтверждения или исключения этой анемии.
Приобретенные гемолитические анемии разделяют на анемии, связанные с воздействием антител, анемии, связанные с соматической мутацией, анемии, связанные с механическим или химическим повреждением эритроцитов, а также обусловленные разрушением эритроцитов паразитами или дефицитом витаминов.
Иммунные гемолитические анемии включают 4 варианта: изоиммунные, трансиммунные, гетероиммунные и аутоиммунные. Изоимунный вариант наблюдается в тех случаях, когда реципиенту перелиты эритроциты и клетки донора, несовместимые по системе АВО. При этом клетки крови донора разрушаются антителами, имеющимися у реципиента и направленными против антигена донора, т.е. гемолизу подвергаются эритроциты донора в крови реципиента (больного). Кроме того, подобная ситуация возможна при антигенной несовместимости клеток крови матери и плода. Трансиммунная гемолитическая анемия развивается у плода, имеющего одинаковые антигены с материнскими, если мать страдает аутоиммунной гемолитической анемией; при этом антитела матери через плаценту проникают в организм плода и оказывают разрушительное действие на его эритроциты. Гетероиммунная гемолитическая анемия отличается от предыдущей тем, что антитела направлены не против эритроцитов, а против антигена, фиксированного на эритроцитах; в ходе реакции антиген-антитело разрушаются и эритроциты. Антитела, к примеру, могут быть направлены против лекарственного препарата, фиксированного на эритроцитах. Гетероиммунной считается также гемолитическая анемия при наличии эритроцитов с измененной антигенной структурой; в этих случаях эритроцит становится «чужим» для организма и иммунная система целенаправленно организует специфическую защиту (синтезируются антитела против измененной антигенной структуры эритроцита).
Об аутоиммунном характере гемолитической анемии свидетельствуют лишь те случаи, когда образующиеся антитела направлены против собственных неизмененных антигенов, конкретно — против нормальной антигенной структуры клеток эритропоэза: эритрокариоцитов или эритроцитов периферической крови. Антитела могут быть представлены неполными тепловыми агглютининами, тепловыми гемолизинами, полными холодовыми агглютининами и двухфазными гемолизинами.
Патогенез аутоиммунной гемолитической анемии. Сущность аутоиммунных процессов состоит в том, что в результате ослабления Т-супрессорной системы иммунитета, контролирующей аутоагрессию, происходит активация В-системы иммунитета, синтезирующей при этом антитела против неизмененных антигенов различных органов. В реализации аутоагрессии принимают участие также Т-лимфоциты-киллеры. Антитела — это иммуноглобулины (Ig), принадлежащие чаще всего к классу G, реже — М и А; они специфичны и направлены против определенного антигена. К IgM относятся, в частности, холодовые антитела и двухфазные гемолизины. Эритроцит, несущий на себе антитела, фагоцитируется макрофагами и в них разрушается; возможен лизис эритроцитов с участием комплемента. Антитела класса IgM могут вызвать агглютинацию эритроцитов непосредственно в кровеносном русле, а антитела класса IgG способны разрушать эритроцит только в макрофагах селезенки. Во всех случаях гемолиз эритроцитов происходит тем интенсивнее, чем больше на их поверхности находится антител. Описана гемолитическая анемия с антителами к спектрину.
Клиника аутоиммунной гемолитической анемии различается в зависимости от вида антител. Чаще других встречается аутоиммунная гемолитическая анемия с неполными тепловыми агглютининами. Заболевание может иметь острое, внезапное начало или сразу же принять хроническое течение. В острых случаях повышается температура тела; кожные покровы более бледны, чем желтушны; могут быть умеренно увеличены периферические лимфатические узлы, селезенка и печень. Анемический синдром сопровождается тахикардией, одышкой, общей слабостью. В общем анализе крови выраженность анемии может быть различной; характер ее обычно нормохромный, умеренно регенераторный; в мазке крови нередко появляются нормобласты, а в период криза наблюдается лейкоцитоз (реже — лейкопения), нередко — тромбоцитопения. В биохимическом анализе крови: умеренная гипербилирубинемия за счет свободной фракции; ОРЭ умеренно снижена. Решающее диагностическое значение имеет положительная прямая проба Кумбса, выявляющая антитела, фиксированные на эритроцитах. При этом следует иметь в виду, что при небольшом количестве антител на эритроцитах, а также после гемолитического криза, когда количество эритроцитов снижается, или после приема глюкокортикоидных гормонов проба Кумбса может быть отрицательной. В этих случаях проводят модифицированную пробу, суть которой состоит в том, что эритроциты 0(1) резус-положительной группы крови наслаивают на агрегированные белки иммунной сыворотки, добавляя их к отмытым эритроцитам больного, — при наличии антител эритроциты склеиваются.
Пароксизмальная холодовая гемоглобинурия и аутоиммунная гемолитическая анемия с полными холодовыми агглютининами характеризуются тем, что у больных антиэритроцитарные антитела проявляют свои агглютинирующие или гемолизирующие свойства при охлаждении. В первую фазу наблюдается фиксация антител на эритроцитах при температуре 0-15 °С; во вторую — наступает внутрисосудистый гемолиз при температуре 37 °С. Холодовые агглютинины в низких титрах присутствуют у 95 % здоровых людей. Холодовые антитела принадлежат к классу IgM; они активируют комплемент крови в дистальных отделах конечностей, так как в них, как правило, температура ниже, чем на других участках тела, — развивается синдром Рейно. Гемолитический криз чаще всего связан с инфекцией микоплазмой (известно, например, что микоплазменная пневмония может осложняться аутоиммунной гемолитической анемией), вирусом Эпштейна-Барр. Клиника соответствует симптоматике внутрисосудистого гемолиза. После гемолитического криза может развиться хроническая форма гемолитической анемии. Первично-хроническая гемолитическая анемия с холодовыми агглютининами развивается иногда у пожилых людей с хроническим лимфолейкозом или парапротеинемическими лейкозами; она носит внутрисосудистый характер. Эритроциты, нагруженные Холодовыми антителами, разрушаются в печени.
Пароксизмальная холодовая гемоглобинурия (синдром Доната-Ландштейнера) характеризуется присутствием антител класса IgG, которые активны в холодной среде и при температуре 37 °С способны активизировать комплемент. Этот вариант гемолитической анемии связан с поздней стадией сифилиса и другими инфекциями, но может быть спровоцирован и локальным охлаждением, например приемом холодной воды. Клиника характерна для внутрисосудистого гемолиза; патогномоничный синдром — боль в поясничной области и моча цвета мясных помоев или темно-коричневой окраски. В моче находят гемоглобин и гемосидерин. Анемия нормохромная, регенераторная; по лабораторным и клиническим данным имеются признаки ДВС-синдрома и почечной недостаточности. В крови увеличено содержание свободной фракции билирубина.
Пароксизмальная ночная гемоглобинурия Маркиафавы—Микели обусловлена приобретенным дефектом мембраны эритроцитов, которая становится чувствительной к нормальному комплементу крови в условиях ацидоза. Дефект мембраны эритроцитов является результатом соматической мутации на ранних этапах клеточной дифференцировки, что подтверждается одновременными тромбоцито- и лейкопенией вследствие неполноценности их мембраны. Клиника пароксизмальной ночной гемоглобинурии характеризуется кризами: они появляются после сна (ночью имеет место физиологический ацидоз), протекают с выраженным болевым синдромом в поясничной области, выделением мочи черного цвета, в которой при исследовании обнаруживают гемосидерин, свободный гемоглобин и железо. Возможны тромботические осложнения: тромбоз периферических вен, воротной вены, вен печени (синдром Бадда-Киари), а также брыжеечных и церебральных вен. Пароксизмальная ночная гемоглобинурия может быть ассоциирована с лейкозами и апластической анемией. Кризы иногда провоцируются инфекцией, приемом препаратов железа. Лабораторные признаки: нормо- или гипохромная анемия, умеренная лейко- и тромбоцитопения. В крови повышен уровень свободного билирубина, содержание сывороточного железа понижено (за счет потери его с мочой, чем обусловлена и гипохромия эритроцитов) или остается в норме. Пробы на кислотную устойчивость эритроцитов (тест Хема) и гемолиз с добавлением глюкозы (сахарозный тест Гармана) выявляют пониженную устойчивость эритроцитов, что патогномонично для пароксизмальной ночной гемоглобинурии.
Гемолитическая анемия, обусловленная механическими факторами, связана с разрушением эритроцитов при их прохождении через измененные сосуды или через искусственные клапаны. Эндотелий сосудов изменяется при васкулитах, злокачественной артериальной гипертензии; при этом адгезия и агрегация тромбоцитов активированы, как и система свертывания крови и образования тромбина. Развиваются распространенный стаз крови и тромбоз мелких кровеносных сосудов (ДВС-синдром) с травматизацией эритроцитов, в результате чего они фрагментируются; в мазке крови находят многочисленные фрагменты эритроцитов (шистоциты). Разрушаются эритроциты также при их прохождении через искусственные клапаны (чаще — при многоклапанной коррекции); описана гемолитическая анемия на фоне сенильного кальцинированного аортального клапана. Диагноз базируется на признаках анемии, повышении концентрации свободного билирубина в сыворотке крови, наличии шистоцитов в мазке периферической крови и симптоматике основного заболевания, ставшего причиной механического гемолиза.
План обследования больного при подозрении на гемолитическую анемию.
План обследования больного можно условно разделить на два этапа:
общие для всех гемолитических анемий лабораторно-инструментальные исследования
Первый этап:
Общий анализ крови с определением количества ретикулоцитов и морфологическим исследованием эритроцитов.
источник
Серповидноклеточная анемия — хроническое гемолитическое заболевание, которые могут вызывать три типа острых нарушений: тяжелая анемия, тяжелые бактериальные инфекции и ишемическую вазоокклюзию вследствие обструкции мелких кровеносных сосудов и капилляров серповидными эритроцитами. Могут возникнуть многочисленные осложнения.
Распространенность носителей серповидных клеток в Европе оценивается примерно в 1/150. В центральной и западной Африке (15-25%) во французской Вест-Индии (10-15%) и в средиземноморских районах (1-15%), высокая распространенность наблюдается также в районах, эндемичных по малярии, так носительство обеспечивает защиту от пернициозной малярии.
Серповидноклеточная анемия является аутосомно-рецессивным заболеванием.
Серповидноклеточная анемия определяется комбинациями двух аномальных аллелей гена бета-глобина, среди которых по крайней мере один несет мутацию beta-6 glu-val (Hb S). Индивидуумы, несущие ген гемоглобина S и гена талассемии бета, подвержены талассодрепаноцитозу. Форма бетаS/betaC (beta-6 glu-lys) также часто встречается.
Наличие фетального гемоглобина объясняет, что болезнь проявляется только через 3 месяца после рождения. Клинические проявления чрезвычайно различны. В дополнение к анемии и бактериальным инфекциям, вазокклюзия вызывают гипералгическую фокальную ишемию, а иногда и инфаркты в мышцах или скелете. С течением времени вазокклюзия могут нарушать целостность тканей или органов.
К другим проявлениям серповидноклеточной анемии относятся бледность, желтуха, ранняя гепатоспленомегалия, атрофия селезенки, повторные кризы, изостенурия, а при длительном течении болезни — некрозы костей, хронические заболевания легких, кардиомегалия и почечная недостаточность.
Диагноз серповидноклеточной анемии основан на анализе гемоглобина с использованием изоэлектрической фокусировки или жидкостной хроматографии высокого давления, теста на растворимость (тест Итано) и молекулярного анализа.
Дифференциальныф диагноз серповидноклеточной анемии включают другие наследственные гемолитические заболевания.
а) Болевой (вазоокклюзионный) криз — самое частое проявление серповидноклеточной анемии. Чаще всего поражаются кости и мышцы. Провоцирующими факторами служат инфекции, дегидратация, холод и гипоксия. Лечение состоит во введении жидкости, в обезболивании и устранении провоцирующего фактора. Легкие приступы, как правило, устраняются приемом анальгетиков и жидкости, более тяжелые требуют госпитализации и в/в инфузии. Так как при длительном течении болезни часто развивается поражение сердца, следует избегать объемной перегрузки. Применение анальгетиков — см. гл. 1, п. III.Б. Седативные средства назначают с осторожностью, поскольку при их передозировке снижается легочная вентиляция. Всем больным серповидноклеточной анемией, получающим большие дозы наркотических анальгетиков, необходим мониторинг дыхания, кровообращения и sO2. При неосложненных болевых кризах переливание эритроцитарной массы не требуется. Следует иметь в виду, что болевые приступы могут имитировать другие осложнения серповидноклеточной анемии, например остеомиелит и холецистит. При боли в груди или животе нужно тщательно наблюдать за больным, так как она может быть первым признаком острого синдрома грудной клетки.
б) Апластический криз чаще всего провоцируется инфекцией, вызванной парвовирусом B19 и приводящей к временному подавлению эритропоэза. Апластический криз характеризуется быстрым снижением гематокрита, исчезновением ретикулоцитов и снижением уровня билирубина в сыворотке. Хотя криз обычно разрешается без вмешательства, анемия может быть столь выраженной, что требуется переливание крови. При серповидноклеточной анемии и других хронических гемолитических анемиях следует избегать контакта с возможными носителями парвовируса — больными инфекционной эритемой или перенесшими апластический криз.
в) Секвестрационный криз обусловлен внезапным скоплением крови в селезенке, реже — в печени. Это угрожающее жизни осложнение серповидноклеточной анемии чаще всего развивается у детей младшего возраста, но может возникать и позднее, особенно при гемоглобинопатии SC, для которой не характерны ранние инфаркты селезенки. Секвестрационный криз проявляется быстрым развитием шока, гепато- или спленомегалии. Лечение состоит в немедленном восполнении ОЦК (см. гл. 3 и гл. 7) и лечении анемии. Поскольку риск рецидива и смертность высоки, после тяжелых кризов проводят спленэктомию. При менее тяжелых кризах (легкая анемия, спленомегалия, нормальная гемодинамика) используют выжидательную тактику. Чтобы отсрочить спленэктомию, назначают постоянные переливания крови, хотя их эффект сомнителен.
г) Гемолитический криз — довольно редкое проявление серповидноклеточной анемии. Ускоренный гемолиз может наблюдаться при сопутствующем дефиците Г-6-ФД, инфекциях или воздействии веществ, вызывающих гемолиз (см. табл. 16.5). При тяжелой анемии показано переливание крови.
д) Мегалобластный криз вызван повышенной потребностью в фолиевой кислоте вследствие усиленного эритропоэза. Его можно предотвратить профилактическим назначением фолиевой кислоты.
е) Гипоксия при серповидноклеточной анемии опасна для жизни, поскольку она способствует внутрисосудистой серповидной деформации эритроцитов, приводящей к инфарктам внутренних органов (головного мозга, легких, почек, печени). У всех детей с любым острым заболеванием следует исключить гипоксемию, особенно если они получают наркотические средства. Чтобы поддержать saO2 на уровне не ниже 98%, назначают кислород; при необходимости прибегают к интубации трахеи и ИВЛ.
ж) Инфекции. Функциональный аспленизм при серповидноклеточной анемии повышает восприимчивость к бактериальным инфекциям, вызванным пневмококками, менингококками, Haemophilus influenzae, сальмонеллами и Escherichia coli. Во всех возрастных группах нередки тяжелые инфекции — пневмония, остеомиелит, сепсис, в том числе уросепсис. Поэтому при лихорадке нужно немедленно обратиться к врачу. Если температура тела выше 38,5°C, показан посев крови и мочи и парентеральное введение антибиотиков (см. гл. 14). Всем детям назначают профилактическую антибиотикотерапию: обычно феноксиметилпенициллин, 125—250 мг 2 раза в сутки. Кроме того, проводят вакцинацию против пневмококков (в возрасте 2 лет) и Haemophilus influenzae (в первые годы жизни).
з) Острый синдром грудной клетки встречается весьма часто. Это основная причина хронических заболеваний легких и смерти у подростков и взрослых с серповидноклеточной анемией. Смерть наступает в результате прогрессирующей дыхательной недостаточности и множественных инфарктов внутренних органов. Синдром проявляется нарушениями дыхания, болью в груди или животе, лихорадкой. Данные рентгенографии грудной клетки в момент возникновения синдрома, как правило, нормальные, но впоследствии часто обнаруживают затемнения. При тяжелом течении поражаются несколько долей, развиваются гипоксемия и анемия. Предрасполагающими факторами служат инфекции верхних дыхательных путей, особенно бактериальные, вирусные и микоплазменные пневмонии. Показано тщательное наблюдение и активное лечение: инфузионная и антимикробная терапия, анальгетики, кислород. В связи с возможностью микоплазменной пневмонии наряду с антибиотиками, активными в отношении наиболее распространенных возбудителей бактериальных инфекций, применяют эритромицин. Эффективность переливания крови окончательно не выяснена, однако в тяжелых случаях острого синдрома грудной клетки применяют обменное переливание с использованием эритроцитарной массы.
и) Инсульт — частое осложнение серповидноклеточной анемии. У детей инсульт обусловлен окклюзией крупных мозговых сосудов, нередко нескольких. Вероятность повторного инсульта высока, но регулярные переливания крови, позволяющие поддерживать гемоглобин S на уровне менее 30%, существенно уменьшают ее. Иногда лечение проводят пожизненно. При острых нарушениях мозгового кровообращения необходимо срочное обменное переливание с использованием эритроцитарной массы.
к) Трансфузионная терапия (см. табл. 16.4) может продлить жизнь, однако она чревата повышением вязкости крови. До тех пор, пока не будет значительно снижен уровень гемоглобина S, гематокрит не должен превышать 25—30%. Неотложное переливание крови показано, когда требуется увеличить транспортную функцию крови без выраженного снижения уровня гемоглобина S. Постоянные переливания крови проводят при необходимости снижения уровня гемоглобина S. Обычно гемоглобин S поддерживают на уровне не более 30%; для этого достаточно вводить эритроцитарную массу в дозе 10—15 мл/кг каждые 3—4 нед. Обменное переливание крови быстро нормализует гематокрит и уровень гемоглобина S. Его проводят по жизненным показаниям. Понижение уровня гемоглобина S необходимо также перед операцией, так как большие операции и общая анестезия могут спровоцировать осложнения, например острый синдром грудной клетки. При серповидноклеточной анемии часто развиваются сенсибилизация к антигенам эритроцитов и трансфузионные реакции. Чтобы предотвратить сенсибилизацию, для переливаний по возможности используют эритроциты, совместимые по дополнительным антигенам — C, D, E, c, e, K. Сенсибилизированным больным переливают только совместимые по дополнительным антигенам препараты крови.
Гидроксикарбамид повышает уровень фетального гемоглобина у больных серповидноклеточной анемией и снижает риск некоторых осложнений при тяжелых форм заболевания.
Трансплантация костного мозга показана в случаях с церебральной васкулопатией.
Прогноз трудно предсказать. Серьезная вазокклюзая или органная недостаточность могут быть причиной смерти.
Синонимы: S-C-гемоглобинопатия, болезнь S-C гемоглобина
Болезнь S-C гемоглобина представляет собой гемоглобинопатию, которая вызывает симптомы, сходные с симптомами серповидноклеточной анемии, но обычно менее выраженными.
Поскольку 10% чернокожих несут признак Hb S (который отвечает за развитие серповидноклеточной анемии), гетерозиготная комбинация S-C встречается чаще, чем гомозиготная гемоглобинопатия Hb C.
Анемия при гемоглобинопатии S-C более мягкая, чем при серповидноклеточной анемии, некоторые пациенты даже имеют нормальные уровни гемоглобина. Однако часто наблюдается гематурия, кровоизлияния в сетчатку и асептический некроз головки бедренной кости. Может также отмечаться спленомегалия.
Лечение как при серповидноклеточной анемии, но определяется выраженностью симптомов.
источник
 Анемия Под определение «гемолитическая анемия» подпадает любая анемия, характеризующаяся преобладанием гемолиза (процессом распада эритроцитов) над эритропоэзом (процессом образования эритроцитов). Гемолиз может быть иммунный и неиммунный. Гемолитическая анемия бывает наследственные и приобретенные.
Анемия Под определение «гемолитическая анемия» подпадает любая анемия, характеризующаяся преобладанием гемолиза (процессом распада эритроцитов) над эритропоэзом (процессом образования эритроцитов). Гемолиз может быть иммунный и неиммунный. Гемолитическая анемия бывает наследственные и приобретенные.
- Наследственная форма гемолитической анемии, обусловленная нарушением мембраны эритроцитов
- Наследственная форма гемолитической анемии, обусловленная нарушением активности ферментов эритроцитов
- Наследственная форма гемолитической анемии, обусловленная нарушением синтеза или структуры гемоглобина
- Анемия, обусловленная влиянием антител
- Анемия, обусловленная изменением структуры мембраны, вызванной соматической мутацией
- Анемия, обусловленная механическим повреждением оболочки эритроцитов
- Анемия, вызванная химическим повреждением эритроцитов
- Анемия, вызванная дефицитом витаминов (фолиевой кислоты и цианокобаламина)
- Анемия, вызванная разрушением эритроцитов паразитами
Болезнь Минковского-Шоффара (наследственный микросфероцитоз) – группа наследственных гемолитических анемий, характеризующихся образованием микросфероцитов (шаровидных эритроцитов) и обусловленных дефектом протеинов цитоскелета эритроцитов. При этом эритроциты теряют часть мембраны, уменьшается соотношение площади поверхности к объему, в результате чего эритроцит превращается в микросфероцит. Как правило, патология наследуется по аутосомно-доминантному признаку. Распространенность наследственного микросфероцитоза составляет примерно 1 случай на 1000-4500 человек.
При наследственном микросфероцитозе генетические нарушения влияют на протеины цитоскелета, преимущественно на те, которые объединяют цитоскелет с мембраной эритроцита. У большинства больных отмечается значительный дефицит спектрина, и только в некоторых случаях этот дефицит обусловлен генетическими дефектами самого спектрина.
Главные признаки наследственного микросфероцитоза – анемия, желтуха, спленомегалия (увеличенная селезенка). Анемия возникает из-за внутриклеточного распада эритроцитов. Желтуха развивается посредством непрямой гипербилирубинемии, может быть непостоянной и, как правило, слабо выражена у детей раннего возраста. Повышенное содержание билирубина в желчи часто является причиной образования пигментных желчных камней (даже у детей). Увеличение селезенки (спленомегалия) отмечается практически во всех случаях. При системных инфекционных патологиях интенсивность гемолиза может увеличиваться, в результате чего развивается спленомегалия.
Тяжелые формы наследственного микросфероцитоза характеризуются деформацией скелета: изменение расположения зубов, акрокефалия (башенный череп), высокое верхнее небо, микрофтальмия (уменьшение глазного яблока). В некоторых случаях отмечаются укороченные мизинцы. Могут образовываться трофические язвы на ногах.
Наследственный микросфероцитоз сопровождается апластическими кризами, которые провоцируются инфекцией (особенно парвовирусной).
Микросфероцитоз – характерное изменение формы эритроцитов при этой патологии. При анализе мазка крови в биологическом материале наблюдаются микросфероциты в виде мелких клеток без центрального просветления (см рисунок 1). Отметим, что обнаружение микросфероцитов в мазках не всегда является признаком наследственного сфероцитоза.
Рисунок 1. Наследственный микросфероцитоз. Микросфероциты в мазке периферической крови (окр. по Романовскому-Гимзе, ув. ×100)
Такой признак обнаруживается при аутоиммунной гемолитической анемии с неполными тепловыми агглютинами, при наследственных дизэритропоэтической анемии. Средний объем эритроцитов, как правило, остается в норме или незначительно снижен. Показатель среднего содержания гемоглобина в эритроцитах в норме или незначительно повышен. Средняя концентрация гемоглобина в эритроцитах повышена почти у 50% пациентов.
Количественным показателем сферичности эритроцитов является осмотическая устойчивость (она снижена). Уровень ретикулоцитов в крови при гемолитическом кризе может значительно повышаться. Миелограмма показывает резкое раздражение красного ростка. Дифференциальный диагноз проводят с аутоиммунной гемолитической анемией, для которой характерна положительная проба Кумбса, отсутствие этой патологии среди родственников пациента и отсутствие данных о начале заболевания в детском возрасте.
Основной метод лечения анемии при наследственном микросфероцитозе – спленэктомия, с помощью которой устраняется анемия; при этом нельзя устранить морфологический дефект эритроцитов.
Наследственная гемолитическая анемия, обусловленная дефицитом глюкозо-6-фосфат дегидрогеназы эритроцитов – наиболее распространенная ферментопатия эритроцитов из группы ферментопатий пентозофосфатного пути метаболизма глюкозы. Глюкозо-6-фосфатдегидрогеназа эритроцитов – олигомер (в зависимости от условий может быть димер или тетрамер), который состоит из субъединиц с молекулярной массой 56 000 D. По данным ВОЗ (Всемирной организации здравоохранения) во всем мире количество людей, страдающих этой патологией, составляет более 200 млн. Наиболее широкое распространение этого заболевания характерно для Средиземноморского региона (Сицилия, Греция, Сардиния), негроидной расы, жителей Ближнего и Дальнего востока.
Клиническая картина при наследственной форме гемолитической анемии полиморфна: степень тяжести патологии может колебаться от гемолитической анемии, возникающей спонтанно после рождения, до гемолитических кризов. Гемолитический криз, который может провоцироваться метаболическим ацидозом или гипогликемией, развивается за несколько часов. В тяжелых случаях у больного развивается гемоглобинурия и шок. Также наблюдаются желтуха, моча приобретает бурый или черный цвет, одышка, диарея, рвота, снижение артериального давления, развивается тяжелая анемия, увеличиваются печень (гепатомегалия) и селезенка (спленомегалия).
Тяжелый гемолитический криз может спровоцировать развитие ДВС-синдрома (диссеминированного внутрисосудистого свертывания крови). Некоторые пациенты не переносят конские бобы (Viciafaba), после употребления которых происходит молниеносное развитие гемолитического криза (это явление также известно, как фовизм или примахиновая анемия).
Дефицит глюкозо-6-фосфат дегидрогеназы эритроцитов необходимо подозревать во всех случаях острого гемолиза, особенно у лиц негроидной расы и жителей средиземноморского региона. Диагноз подтверждается путем проведения лабораторных анализов. Острый гемолиз характеризуется быстрым снижением гематокрита с одновременным повышением уровня гемоглобина и непрямого гемоглобина, а также снижением уровня гаптоглобина. Анализ мазка крови показывает наличие фрагментов эритроцитов. Основой диагностики считается качественное (при необходимости – количественное) определение активности глюкозо-6-фосфат дегидрогеназы эритроцитов. У пациентов с вариантом «А-» явление аномального гемолиза проходит, как правило, самостоятельно – такие больные не нуждаются в специальном лечении. В случае развития тяжелого гемолитического криза необходимо проводить форсированный диурез, профилактику ДВС-синдрома, плазмаферез (с целью удаления продуктов гемолиза).
В случае возникновения качественной гемоглобинопатии происходит изменение аминокислотной последовательности цепей глобина. Талассемия (количественная гемоглобинопатия) характеризуется снижением образования цепей глобина без изменения их цепей. Нужно отметить, что разница между качественной и количественной гемоглобинопатиями не абсолютна.
Талассемия (анемия Кули) – группа патологий, обусловленных генетическим нарушением синтеза одной из цепей глобина. В норме процесс синтеза глобиновых цепей сбалансирован, поэтому свободных цепей глобина нет. В случае нарушения синтеза одной из цепей глобина баланс нарушается, образуются лишние цепи, которые агрегируют и откладываются в эритрокариоцитах. Среди жителей Средиземноморья наиболее распространена β-талассемия.
«Большая талассемия» (болезнь Кули, β-талассемия) – наследственная гемолитическая анемия, впервые описанная американскими педиатрами-гематологами Томасом Бентоном Кули (Thomas Benton Cooley) и Ли (P. Lee) в статье «Серия случаев спленомегалии у детей с анемией и необычными изменениями костей» («A Series of Cases of Splenomegaly in Children, with Anemia and Peculiar Bone Changes»), где были приведены случаи у выходцев из стран Средиземноморья. Для анемии Кули характерна тяжелая степень течения с самого детства, задержка роста и изменения костей в результате увеличения объема костного мозга, возникающие в случае отсутствия соответствующего лечения). Также при этой патологии у больного наблюдаются гепатомегалия, спленомегалия, гиперспленизм, деформации черепа (монголоидное лицо, башенный череп); желтуха, бледность и отложение меланина придают коже особый медный оттенок. Кроме этого, наблюдается перегрузка железом сердца, легких, печени, поджелудочной железы и других органов эндокринной системы, переломы костей, сдавления периферических нервов, разного рода инфекционные осложнения.
Результаты лабораторных исследований периферической крови показывают гипохромную анемию, ретикулоцитоз, мишеневидные эритроциты (см рис 2-4).
Рисунок 02. Анемия Кули (большая талассемия). Периферическая кровь. Микроцитоз, выраженная гипохромия, мишеневидные нормобласты и эритроциты (окр. по Романовскому-Гимзе, ув. ×100)
Рисунок 03. Анемия Кули (большая талассемия). Периферическая кровь (окр. по Романовскому-Гимзе, ув. ×50)
Рисунок 04. Анемия Кули (большая талассемия). Периферическая кровь. Множественные мишеневидные эритроциты (окр. по Романовскому-Гимзе, ув. ×100)
Миелограмма демонстрирует раздражение «красного ростка» и повышение количества сидеробластов. Также наблюдается повышение осмотической резистентности эритроцитов и количества билирубина за счет непрямой фракции. В крови повышается содержание железа и ферритина, развивается гемосидероз (чрезмерное отложение гемосидерина в тканях) внутренних органов. При гомозиготной β-талассемии необходимо проводить пренатальную диагностику – забор клеток плода из амниотической жидкости на предмет выявления мутации генов, отвечающих за кодирование β-цепи глобина, с применением метода полимеразной цепной реакции.
Без соответствующего лечения больные анемией Кули умирают в детском возрасте. Продлить жизнь, предупредить деформации костей и задержку роста можно путем регулярных трансфузий эритроцитарной массы (лучше переливать отмытые или размороженные эритроциты) при условии поддержания достаточно высокого уровня гемоглобина. В случае значительной спленомегалии и явлениях гиперспленизма больному показана спленэктомия (удаление селезенки). С целью предотвращения развития гемосидероза пациентам периодически назначают Деферазирокс (Эксиджад) или Дефероксамин (Десферал). Излечение возможно при аллогенной трансплантации костного мозга.
Серповидноклеточная анемия обусловлена носительством гемоглобина, который меняет свою структуру в условиях гипоксии. Самой распространенной аномалией структуры гемоглобина является гемоглобинопатия Sα2β26 глу+вал. При гомозиготном носительстве можно говорить о серповидноклеточной анемии; при гетерозиготном носительстве – серповидноклеточная аномалия. Патология наследуется по аутосомно-доминантному признаку. При серповидноклеточной анемии наблюдается мутация, в результате которой в цепи глобина глутаминовая кислота заменяется валином. В результате растворимость гемоглобина S при отдаче кислорода снижается, что приводит к образованию геля.
Серповидноклеточная анемия наиболее распространена среди населения Центральной Африки, Турции, Индии, Кубы. У больных диагностируется анемия, тромботические осложнения, поражения костей и суставов (отмечаются некрозы плечевой и бедренной костей). Кроме этого, тромбозы осложняются инфарктами (сердца, легких, почек, селезенки, головного мозга), приступами сильной боли в области живота. У детей отмечаются нарушения физического (отставание в росте) и полового развития, ночное недержание мочи, нарушение зрения (тромбозы сосудов сетчатки). Также могут развиваться гемолитический, апластический и секвестрационные кризы, при этом в селезенке происходит резкое накопление эритроцитов, что вызывает гиповолемический шок и резкое снижение уровня гемоглобина.
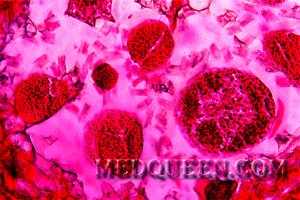
Для анализов крови при апластической анемии характерны низкий уровень гемоглобина, наличие серповидных эритроцитов (рисунок 5), базофильная пунктация эритроцитов, их мишеневидность, повышение уровня ретикулоцитов и непрямого билирубина. Миелограмма демонстрирует раздражение «красного ростка».
Рисунок 5. Серповидноклеточная анемия. Периферическая кровь. Серповидные и мишеневидные эритроциты. выраженная гипохромия эритроцитов (окр. по Романовскому-Гимзе, ув. ×100)
В качестве лечения применяют адекватную инфузионную терапию, переливания эритроцитарной массы, оксигенотерапии.
К приобретенным гемолитическим анемиям относится группа заболеваний разного патогенеза, которые объединяет внутрисосудистый гемолиз (гемолиз эритроцитов в периферической крови). В зависимости от механизма эритролиза приобретенная гемолитическая анемия может носить иммунный и неиммунный характер. Но, несмотря на разные патогенетические механизмы, клинические признаки этих анемий часто совпадают.
Гемолитическая анемия у пациентов с протезированными клапанами сердца и сосудами развивается примерно в 10% случаев при протезированном аортальном клапане. При использовании стеллитовых запирательных элементов частота гемолиза незначительно увеличивается (по сравнению с селиконовыми). Также некоторое увеличение частоты гемолиза отмечается при наличии околоклапанной регургитации и при малом диаметре клапана. Биопротезы (свиные клапаны) в редких случаях являются причиной механического гемолиза. Гораздо реже причиной гемолиза может быть также протезированный митральный клапан, так как трансклапанный градиент давления в этом случае ниже.
Гемолиз протезированными клапанами происходит в результате одновременного действия сразу нескольких факторов:
- Значительная сила сдвига, которая при турбулентном токе крови действует на мембрану эритроцитов, особенно когда под высоким давлением кровь проходит через маленькое отверстие (например, при околоклапанной регургитации)
- Отложения фибрина на участках неплотного прилегания кольца клапана к тканям сердца
- Прямое механическое повреждение эритроцитов при закрытии запирательного элемента
Значительное разрушение эритроцитов может наблюдаться после закрытия дефекта межпредсердной перегородки типа ostium primum заплатой из синтетического материала. Умеренное сокращение жизни эритроцитов с легкой анемией или без нее может наблюдаться при значительном обызвествлении аортального клапана. Механический гемолиз обнаруживается также у пациентов, перенесших аортокоронарное и аортобедренное шунтирование.
Тяжелые случаи механического гемолиза сопровождаются тяжелой анемией, ретикулоцитозом, обнаруживаются фрагментированные эритроциты (шизоциты), гемоглобинемия и гемоглобинурия, повышается активность лактатдегидрогеназы, снижается уровень гаптоглобина. Выведение железа из организма с мочой в виде гемосидерина или гемоглобина может вызвать дефицит железа в организме. В случае развития дефицита железа пациенту назначается пероральный прием препаратов железа. Терапия препаратами железа способствует повышению уровня гемоглобина и способствует снижению сердечного выброса и снижению интенсивности гемолиза. Отметим, что ограничение физической активности также способствуют снижению интенсивности распада эритроцитов. Если предпринимаемые меры не приводят к желаемому результату, нужно полностью устранить околоклапанную регургитацию или заменить протез.
источник
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Серповидно-клеточная анемия протекает в виде эпизодов болевых приступов (кризов), связанных с окклюзией капилляров в результате спонтанного «серпления» эритроцитов, чередующихся с периодами ремиссии. Кризы могут провоцироваться интеркуррентными заболеваниями, климатическими условиями, стрессами, возможно спонтанное возникновение кризов.
 [1], [2], [3], [4], [5], [6]
[1], [2], [3], [4], [5], [6]
Симптомы серповидно-клеточной анемии обычно появляются к концу первого года жизни. У новорожденных детей преобладает фетальный гемоглобин (HbF), по мере снижения в постнатальном периоде HbF растет концентрация HbS. Внутрисосудистое «серпление» и признаки гемолиза могут обнаруживаться уже в возрасте 6-8 нед, однако клинические проявления заболевания, как правило, не характерны до 5-6-месячного возраста.
Больные серповидно-клеточной анемией имеют типичный только для этого заболевания внешний вид: удлиненный нижний сегмент тела, дорсальный кифоз и люмбальный лордоз, готическое небо, выступающий лоб, башенный череп, значительное удлинение конечностей, которое зависит от замедления процессов окостенения в эпифизах, общей задержки созревания костей. Характерно отставание в физическом и половом развитии. В возрасте до 2 лет показатели физического развития соответствуют норме, затем в возрасте 2-6 лет рост и вес значительно замедляются, причем отставание в весе выражено в большей степени, чем в росте. К концу подросткового периода больные дети обычно догоняют здоровых по росту, отставание в весе сохраняется. Отмечается задержка полового созревания, у мальчиков пубертатный возраст наступает в 16-18 лет, у девочек — в 15-17 лет. Уровень интеллектуального развития у больных нормальный.
У всех больных наблюдается бледность кожи и слизистых, желтушность, усиливающаяся с возрастом. Начиная с 6 мес жизни у больных пальпируется селезенка, в начале заболевания размеры селезенки увеличены значительно, на более поздних этапах, вследствие развития фиброза на фоне повторных инфарктов, селезенка уменьшается в размерах (аутоспленэктомия) и у детей старше 6 лет спленомегалию обнаруживают редко. Хотя размеры селезенки в начале заболевания значительно увеличены, клинически отмечается функциональный гипоспленизм. Лабораторно при функциональном гипоспленизме в периферической крови возможен транзиторный тромбоцитоз, в эритроцитах обнаруживают тельца Жолли. У больных с аутоспленэктомией появляются мишеневидные клетки и акантоциты. У части детей имеется гепатомегалия. Нередко выявляется кардиомегалия. Характерна аденопатия, инволюция миндалин у таких детей происходит медленно. У больных уже к возрасту 3-4 лет возможно развитие желчнокаменной болезни, частота холелитиаза у больных в возрасте 2-4 лет составляет 12 %, в возрасте 1 5 — 1 8 лет — 42 % ; довольно часто встречается язвенная болезнь двенадцатиперстной кишки.
Заболевание протекает хронически, больные тяжелой формой серповидно-клеточной анемии живут около 20 лет. Периодически отмечаются острые состояния — кризы. Различают два типа кризов: клинические (болевые или вазоокклюзионные), при которых показатели состава гемоглобина и ретикулоцитов в основном не отличаются от нормы; гематологические, с резким снижением уровня гемоглобина и ретикулоцитозом. Нередко кризы сочетаются.
Клинические кризы (болевые, вазоокклюзионные, ревматоидные и абдоминальные) являются наиболее частым вариантом серповидно-клеточной анемии. Могут быть спровоцированы инфекциями или возникают спонтанно. Болевой синдром связан с возникновением инфарктов вследствие окклюзии сосудов серповидными эритроцитами. Инфаркты могут быть в костном мозге, костях и надкостнице, периартикулярных тканях суставов. Основной признак вазоокклюзионных кризов — боль различной интенсивности, сопровождающаяся температурной реакцией, отеком в области поражения, воспалительной реакцией. Первым проявлением заболевания в грудном возрасте может стать симметричное болезненное опухание кистей рук и стоп (вследствие окклюзии плюсневых и пястных костей) — серповидно-клеточный дактилит. Рентгенологически выявляется деструкция костной ткани, сопровождающаяся периостальной реакцией. У больных старшего возраста отмечаются болезненность и опухание крупных суставов и окружающих их тканей. Инфаркты анатомических структур, располагающихся в полости живота, приводят к возникновению абдоминальных болей, напоминающих клинику острого живота. Серьезную опасность представляют острые неврологические нарушения, отмечающиеся примерно у 25 % больных, в том числе припадки, тромботические и геморрагические инсульты, транзиторные ишемические приступы. Мозговые инсульты — результат окклюзии крупного сосуда, возникают в основном у детей (примерно у 7 % больных; частота в среднем составляет 1,7 % в год в течение первых 20 лет жизни, причем частота инсультов максимальна у детей в возрасте 5-10 лет), могут оставлять после себя необратимые последствия в виде гемиплегий и в 70 % случаев при отсутствии лечения рецидивируют в течение 3 лет. У взрослых больных возможно возникновение острых геморрагических инсультов в результате неоваскуляризации и образования аневризм сосудов головного мозга. Развиваются инфаркты легких, которые трудно отдифференцировать от пневмонии, у больного отмечаются одышка, кровохарканье. У детей острый торакальный синдром отличается более тяжелым течением и является самой частой причиной гибели больных. Смерть наступает в результате прогрессирующей дыхательной недостаточности и множественных инфарктов внутренних органов. Острый торакальный синдром вызван появлением в микрососудистом русле легких серповидных клеток и проявляется нарушениями дыхания, болью в грудной клетке или животе, лихорадкой. Данные рентгенографии грудной клетки в момент возникновения синдрома, как правило, нормальные, но впоследствии часто обнаруживают инфильтраты (при тяжелом течении поражаются несколько долей). В 50 % случаев предрасполагающими факторами являются инфекции верхних дыхательных путей, вызванные Streptococcus pneumoniae, Mycoplasma, Chlamydia; в 15 % случаев причиной развития ОТС может быть жировая эмболия легких. В костном мозге возникают некрозы, инфаркты, развивается жировая эмболия, для которой характерны лихорадка, беспокойство, тревога, расстройство сознания, кома и другие нарушения психоневрологического статуса. Могут отмечаться тромбоцитопения и клиническая картина ДВС-синдрома. Большое значение придается исследованию глазного дна — жировые эмболы обнаруживаются в сосудах сетчатки. Проявлением вазоокклюзионного криза является также острая патология мочеполовой системы. Рецидивирующий приапизм наблюдается более чем у 50 % мужчин с серповидно-клеточной анемией. Предрасполагающими к возникновению приапизма факторами являются половой акт, мастурбация, инфекции, локальная травма. Лечение приапизма должно быть начато в течение первых 12 часов, назначают заменные переливания крови с целью уменьшения эрекции, предотвращения рубцевания и развития импотенции. Если консервативное лечение неэффективно, прибегают к хирургическому вмешательству, обеспечивающему декомпрессию пещеристых тел. Появление серповидных клеток в мозговом слое почки обусловливает возникновение некроза почечных сосочков и гематурии. Окклюзия серповидными эритроцитами сосудов печени проявляется болевым синдромом, симулирующим острый холецистит или вирусный гепатит, выраженной гепатомегалией, резким нарастанием билирубина (главным образом, прямого) и активности аминотрансфераз. Возможны фулминантная печеночная недостаточность, массивный холестаз, развитие энцефалопатии и шока, что требует заменного переливания крови.
У больных серповидно-клеточной анемей выявляют изменения в системе гемостаза. Отмечаются гиперкоагуляция, выраженная внутрисосудистая активация и агрегация тромбоцитов, повышение уровня фактора Виллебранда, повышение концентрации фибриногена, дефицит протромбинов С и S, что значительно увеличивает риск возникновения тромбозов. Изменения в системе гемостаза имеют значение в генезе вазоокклюзионных кризов.
Вазоокклюзивный (болевой) криз
Самое частое проявление серповидноклеточной анемии. В основном поражаются кости и мышцы. Провоцирующими факторами служат инфекции, дегиратация, холод и гипоксия. Дактилит (синдром рук-ног) — болезненное опухание тыльных поверхностей кистей и стоп — характерен для детей до 5 лет. Костные, клинически сходные с остеомиелитом, чаще начинаются в возрасте 3-4 лет. Абдоминальные симптомы (опоясывающий синдром) развиваются в результате окклюзии сосудов брыжейки и инфаркта печени, селезёнки или лимфатических узлов, в этих случаях необходима дифференциальная диагностика с острым животом. Лёгочный синдром (острый грудной синдром) встречается достаточно часто, в основном у подростков и взрослых, и служит основной причиной хронических заболеваний лёгких и смерти, наступающей в результате прогрессирующей дыхательной недостаточности и множественных инфарктов внутренних органов. Острый грудной синдром необходимо дифференцировать с пневмонией. Лечение симптоматическое (антибактериальная и инфузионная терапия, анальгетики, кислород). Такие факторы, как половая связь, мастурбация, инфекция и локальная травма, способствуют развитию приапизма, в некоторых случаях приводящего к импотенции. Болезненная гематурия умеренной степени развивается вследствие папиллярного некроза почек. Кризы ЦНС могут сопровождаться:
- судорогами;
- менингеальными признаками;
- слепотой;
- ретинопатией;
- головокружением;
- острым нарушением мозгового кровообращения;
- инфарктом мозга.
Частота встречаемости кризов ЦНС составляет 7-29%, средний возраст их развития — 7,7 лет. Высок риск развития субарахноидальных кровоизлияний.
Наиболее часто локализуется в селезёнке (селезёночная секвестрация), развивается редко, в возрасте 5-24 мес, часто приводит к летальному исходу. Признаки, характерные для клинической картины:
- спленомегалия (сброс большого количества крови в селезёнку);
- внезапная острая боль в животе, сопровождающаяся тошнотой и рвотой;
- резкое снижение уровня Нb, приводящее к гиповолемическому шоку и смерти.
Печёночная секвестрация проявляется:
- внезапным болезненным увеличением печени;
- выраженным повышением уровня билирубина за счёт его прямой фракции;
- повышением активности трансаминаз (АЛТ, ACT).
Может развиться в любом возрасте при фиброзе селезёнки. Лечение состоит в немедленном восполнении ОЦК и коррекции анемии, а также удалении селезёнки.
Наиболее часто вызывается парвовирусной инфекций В19. Признаки, характерные для клинической картины:
- резкое глубокое снижение уровня гемоглобина (вплоть до 10 г/л) с отсутствием ретикулоцитов и нормобластов в периферической крови;
- число тромбоцитов и лейкоцитов, как правило, не изменено;
- существенное снижение уровня билирубина в сыворотке крови.
Купируется чаще всего самостоятельно в течение 10 дней. При резком снижении содержания Нb показано переливание эритроцитарной массы.
Сопровождается резкой слабостью, бледностью, иктеричностью склер, могут быть боли в животе. В общем анализе крови отмечается снижение гематокрита до 15% и ниже, ретикулоцитоз. Через несколько дней гемолиз постепенно прекращается. При тяжёлой анемии показано переливание эритроцитарной массы.
Частое осложнение серповидноклеточной анемии у детей. Развивается вследствие окклюзии крупных сосудов головного мозга, часто нескольких. Вероятность повторных инсультов высока. Регулярные переливания эритроцитарной массы, поддерживающие уровень Hb S не более 30%, существенно снижают риск развития повторных инсультов. При острых нарушениях мозгового кровообращения необходимы срочное обменное переливание с использованием эритроцитарной массы, дегидратация с защелачиванием.
Вызван повышенной потребностью в фолиевой кислоте в результате усиленного эритропоэза, предотвращается профилактическим приёмом фолиевой кислоты внутрь.
При серповидно-клеточной анемии за счет повторяющихся вазоокклюзионных кризов и хронического гемолиза отмечаются выраженные хронические изменения во многих органах. Нарушения со стороны сердца проявляются тахикардией, одышкой. Сердце устойчиво к окклюзионным поражениям вследствие того, что сокращение миокарда облегчает прохождение дефектных эритроцитов по сосудам, питающим орган, и это предотвращает развитие тромбов. Однако в результате постоянной гипоксии (хроническая анемия) развивается кардиомегалия, постепенно прогрессируют вторичный фиброз и гемосидероз миокарда. При обследовании по результатам ЭКГ обнаруживают синусовую тахиаритмию, левограмму, гипертрофию левого желудочка, инверсию зубца Т; рентгенологически отмечается увеличение всех полостей сердца, выбухание легочной артерии; при эхокардиографии выявляется дилатация и левого, и правого желудочков. У больных старшего возраста развивается легочная гипертензия и легочное сердце. Рецидивирующие легочные инфаркты у ряда больных становятся причиной фиброза легких. Возникновение ацидоза и гиперосмолярности мозгового слоя почек связано с образованием серповидных клеток, поэтому у всех больных серповидно-клеточной анемией рано возникает хроническая патология почек. Почки вследствие ишемии поражаются вторичным гломерулонефритом, диффузный фиброз канальцев и клубочков почек приводит к прогрессирующему ухудшению функции почек (первым проявлением облитерации сосудов мозгового слоя почек является гипостенурия, обнаруживаемая уже к 10 годам жизни); нарушение концентрационной способности почек делает больных с серповидно-клеточной анемией особенно чувствительными к дегидратации. Дефекты канальцев могут проявляться в виде тубулярного ацидоза и гиперкалиемии. В некоторых случаях наблюдается нефротический синдром. Поражение печени проявляется хронической гепатомегалией; зоны некроза в печени в последующем фиброзируются, гепатопатия может перейти в цирроз. Возможно развитие посттрансфузионного гепатита. Вследствие окклюзии сосудов мозга отмечаются неврологические нарушения: дефекты речи, нарушения походки, гемипарезы. Нередки поражения глаз с осложнениями в виде отслойки сетчатки. Развивающиеся патологические процессы зависят от локализации поражения. У младших детей из-за развития анастомозов в подкожной клетчатке поражение кожи (трофические язвы нижних конечностей) не встречается, у старших детей и взрослых нарушения кровоснабжения могут вызывать некроз кожи. Функциональный гипоспленизм при серповидно-клеточной анемии повышает восприимчивость к бактериальным инфекциям, вызванным пневмококками, менингококками, Н. Influenzae, сальмонеллами и Е. Coli. Во всех возрастных группах нередки тяжелые инфекции — пневмония, менингит, остеомиелит, сепсис, в том числе уросепсис. Периодом максимального риска гибели от тяжелых инфекций являются первые 5 лет жизни.
[7], [8], [9], [10], [11], [12], [13], [14]
источник
Серповидно-клеточная анемия – наследственная гемоглобинопатия, обусловленная синтезом аномального гемоглобина S, изменением формы и свойств эритроцитов крови. Серповидно-клеточная анемия проявляется гемолитическими, апластическими, секвестрационными кризами, тромбозами сосудов, костно-суставными болями и припухлостью конечностей, изменениями скелета, сплено- и гепатомегалией. Диагноз подтверждается по данным исследования периферической крови и пунктата костного мозга. Лечение серповидно-клеточной анемии является симптоматическим, направленным на предупреждение и купирование кризов; может быть показано переливание эритроцитов, прием антикоагулянтов, проведение спленэктомии.
Серповидно-клеточная анемия (S-гемоглобинопатия) – разновидность наследственной гемолитической анемии, характеризующаяся нарушением структуры гемоглобина и присутствием в крови эритроцитов серповидной формы. Заболеваемость серповидно-клеточной анемией распространена, главным образом, в странах Африки, Ближнего и Среднего Востока, Средиземноморского бассейна, Индии. Здесь частота носительства гемоглобина S среди коренного населения может достигать 40%. Любопытно, что больные серповидно-клеточной анемией имеют повышенную врожденную устойчивость к заражению малярией, поскольку малярийный плазмодий не может проникнуть в эритроциты серповидной формы.
В основе серповидно-клеточной анемии лежит генная мутация, обусловливающая синтез аномального гемоглобина S (HbS). Дефект структуры гемоглобина характеризуется заменой глутаминовой кислоты валином в ß-полипептидной цепи. Образующийся при этом гемоглобин S после потери присоединенного кислорода приобретает консистенцию высокополимерного геля и становится в 100 раз менее растворимым, чем нормальный гемоглобин А. В результате этого эритроциты, несущие деоксигемоглобин S, деформируются и приобретают характерную полулунную (серповидную) форму. Измененные эритроциты становятся ригидными, малопластичными, могут закупоривать капилляры, вызывая ишемию тканей, легко подвергаются аутогемолизу.
Наследование серповидно-клеточной анемии происходит по аутосомно-рецессивному типу. При этом, гетерозиготы наследуют дефектный ген серповидно-клеточной анемии от одного из родителей, поэтому, наряду с измененными эритроцитами и HbS, имеют в крови и нормальные эритроциты с HbА. У гетерозиготных носителей гена серповидно-клеточной анемии признаки заболевания возникают лишь в определенных условиях. Гомозиготы наследуют по одному дефектному гену от матери и от отца, поэтому в их крови присутствуют только серповидные эритроциты с гемоглобином S; заболевание развивается рано и протекает тяжело.
Таким образом, в зависимости от генотипа, в гематологии различают гетерозиготную (HbAS) и гомозиготную (HbSS, дрепаноцитоз) форму серповидно-клеточной анемии. К редко встречающимся вариантам заболевания относятся промежуточные формы серповидно-клеточной анемии. Обычно они развиваются у двойных гетерозигот, несущих один ген серповидно-клеточной анемии и другой дефектный ген — гемоглобина C (HbSC), серповидной β-плюс (HbS/β +) или β-0 (HbS/β0) талассемии.
Гомозиготная серповидно-клеточная анемия обычно проявляется у детей к 4-5 месяцу жизни, когда увеличивается количество HbS, а процентное содержание серповидных эритроцитов достигает 90%. В таких случаях раннее возникновение гемолитической анемии у ребенка обуславливает задержку физического и умственного развития. Характерны нарушения развития скелета: башенный череп, утолщение лобных швов черепа в виде гребня, кифоз грудного или лордоз поясничного отдела позвоночника.
В развитии серповидно-клеточной анемии выделяют три периода: I — с 6 месяцев до 2-3 лет, II — с 3 до 10 лет, III — старше 10 лет. Ранними сигналами серповидно-клеточной анемии служат артралгии, симметричное опухание суставов конечностей, боли в груди, животе и спине, желтушность кожи, спленомегалия. Дети с серповидно-клеточной анемией относятся к категории часто болеющих. Степень тяжести течения серповидно-клеточной анемии тесно коррелирует с концентрацией HbS в эритроцитах: чем она выше, тем тяжелее выражена симптоматика.
В условиях интеркуррентной инфекции, стрессовых факторов, обезвоживания, гипоксии, беременности и пр. у больных данным видом наследственной анемии могут развиваться серповидно-клеточные кризы: гемолитический, апластический, сосудисто-окклюзионный, секвестрационный и др.
При развитии гемолитического криза состояние больного резко ухудшается: возникает фебрильная лихорадка, в крови повышается непрямой билирубин, усиливается желтушность и бледность кожных покровов, появляется гематурия. Стремительный распад эритроцитов может привести к анемической коме. Апластические кризы при серповидно-клеточной анемии характеризуются угнетением эритроидного ростка костного мозга, ретикулоцитопенией, снижением гемоглобина.
Следствием депонированием крови в селезенке и печени служат секвестрационные кризы. Они сопровождаются гепато- и спленомегалией, сильными болями в животе, резкой артериальной гипотонией. Сосудисто-окклюзионные кризы протекают с развитием тромбоза сосудов почек, ишемии миокарда, инфаркта селезенки и легких, ишемического приапизма, окклюзии вен сетчатки, тромбоза мезентериальных сосудов и др.
Гетерозиготные носители гена серповидно-клеточной анемии в обычных условиях ощущают себя практически здоровыми. Морфологически измененные эритроциты и анемия у них возникают только в ситуациях, связанных с гипоксией (при тяжелой физической нагрузке, авиаперелетах, восхождении в горы и др.). Вместе с тем, остро развившийся гемолитический криз при гетерозиготной форме серповидно-клеточной анемии может иметь летальный исход.
Хроническое течение серповидно-клеточной анемии с повторными кризами приводит к развитию целого ряда необратимых изменений, нередко становящихся причиной гибели больных. Примерно у трети больных отмечается аутоспленэктомия – сморщивание и уменьшение размеров селезенки, вызванное замещением функциональной ткани рубцовой. Это сопровождается изменением иммунного статуса больных серповидно-клеточной анемией, более частым возникновением инфекций (пневмонии, менингита, сепсиса и др.).
Исходом сосудисто-окклюзионных кризов могут стать ишемические инсульты у детей, субарахноидальные кровоизлияния у взрослых, легочная гипертензия, ретинопатия, импотенция, почечная недостаточность. У женщин с серповидно-клеточной анемией отмечается позднее становление менструального цикла, склонность к самопроизвольному прерыванию беременности и преждевременным родам. Следствием ишемии миокарда и гемосидероза сердца служит возникновение хронической сердечной недостаточности; повреждения почек — хронической почечной недостаточности.
Длительный гемолиз, сопровождаемый избыточным образованием билирубина, приводит к развитию холецистита и желчнокаменной болезни. У больных серповидно-клеточной анемией часто возникают асептические некрозы костей, остеомиелит, язвы голеней.
Диагноз серповидно-клеточной анемии выставляется гематологом на основании характерных клинических симптомов, гематологических изменений, семейно-генетического исследования. Факт наследования ребенком серповидно-клеточной анемии может быть подтвержден еще на этапе беременности с помощью биопсии ворсин хориона или амниоцентеза.
В периферической крови отмечается нормохромная анемия (1-2х1012/л), снижение гемоглобина (50-80 г/л), ретикулоцитоз (до 30%). В мазке крови обнаруживаются серповидно измененные эритроциты, клетки с тельцами Жолли и кольцами Кабо. Электрофорез гемоглобина позволяет определить форму серповидно-клеточной анемии – гомо- или гетерозиготную. Изменение биохимических проб крови включает гипербилирубинемию, увеличение содержания сывороточного железа. При исследовании пунктата костного мозга выявляется расширение эритробластического ростка кроветворения.
Дифференциальная диагностика направлена на исключение других гемолитических анемий, вирусного гепатита А, рахита, ревматоидного артрита, туберкулеза костей и суставов, остеомиелита и др.
Серповидно-клеточная анемия относится к категории неизлечимых болезней крови. Таким пациентам требуется пожизненное наблюдение гематолога, проведение мероприятий, направленных на предупреждение кризов, а при их развитии – проведение симптоматической терапии.
В период развития серповидно-клеточного криза требуется госпитализация. С целью быстрого купирования острого состояния назначается кислородотерапия, инфузионная дегидратация, введение антибиотиков, обезболивающих средств, антикоагулянтов и дезагрегантов, фолиевой кислоты. При тяжелом течении обострений показано переливание эритроцитарной массы. Проведение спленэктомии не способно повлиять на течение серповидно-клеточной анемии, однако может на время уменьшить проявления заболевания.
Прогноз гомозиготной формы серповидно-клеточной анемии неблагоприятный; большая часть пациентов погибает в первое десятилетие жизни от инфекционных или тромбоокклюзионных осложнений. Течение гетерозиготных форм патологии гораздо более обнадеживающее.
Для предупреждения быстро прогрессирующего течения серповидно-клеточной анемии следует избегать провоцирующих условий (обезвоживания, инфекций, перенапряжения и стрессов, экстремальных температур, гипоксии и пр.). Детям, страдающим данной формой гемолитической анемии, в обязательном порядке показана вакцинация против пневмококковой и менингококковой инфекции. При наличии в семье больных серповидно-клеточной анемией необходима медико-генетическая консультация для оценки риска развития заболевания у потомства.
источник