
Анемия Даймонда-Блекфена — самая известная форма парциальной красноклеточной аплазии у детей. Заболевание названо по имени авторов, описавших в 1938 г. четверых детей с характерными признаками заболевания.
Всего зарегистрировано более 500 случаев анемии Даймонда-Блекфена, частоту синдрома оценивают как 4-10 случаев на 1 000 000 родившихся, соотношение мальчиков и девочек составляет около 1:1. Семейные случаи составляют 10-20% всех случаев анемии Даймонда-Блекфена, в том числе заболевание было диагностировано у монозиготных близнецов. Доказан как аутосомно-доминантный, так и аутосомно-рецессивный путь наследования. 80-90% случаев анемии Даймонда-Блекфена диагностируют в течение первого года жизни, причём у 25% пациентов анемию выявляют уже на момент рождения. Диагноз анемии Даймонда-Блекфена у детей старшего возраста должен ставиться с осторожностью, после исключения приобретённых форм ПККА. Примерно 25-30% случаев анемии Даймонда-Блекфена связано с мутацией гена рибосомального белка S19, значение которого для эритропоэза неизвестно. Ещё одним хромосомным локусом, связанным с развитием заболевания, является 8р22-р23.
 [1], [2], [3], [4]
[1], [2], [3], [4]
Анемия Даймонда-Блекфена — наследственное заболевание с предположительно аутосомно-рецессивным типом наследования, с одинаковой частотой среди больных встречаются мальчики и девочки. Среди механизмов развития заболевания указывают на аномалию эритроидных клеток-предшественников, дефект их микроокружения в костном мозге, клеточно-опосредованную супрессию и наличие гуморальных ингибиторов эритропоэза. К постоянным признакам болезни относят уменьшение числа эритроидных единиц в костном мозге, повышение уровня эритропоэтинов в крови, дефект добавочных клеток костного мозга.
[5], [6], [7], [8], [9], [10], [11], [12]
Симптомы исчерпывается бледностью и другими симптомами тяжёлой анемии. Увеличение печени и селезёнки не характерно для заболевания, однако в дальнейшем в результате формирования фиброза и/или цирроза печени в результате перегрузки железом и течения посттрансфузионных гепатитов В и С гепатоспленомегалия становится типичным симптомом.
Для больных анемией Даймонда-Блекфена характерны врождённые аномалии развития, однако их спектр и выраженность значительно отличаются от анемии Фанкони. Характерно также хроническое течение анемии Даймонда-Блекфена; у части больных, чаще в период пубертата, отмечают спонтанную ремиссию. Анемия Даймонда-Блекфена — предлейкемический синдром: ОМЛ развились минимум у 8 больных.
Диагностические критерии анемии Даймонда-Блекфена:
- нормохромная, часто макроцитарная анемия;
- глубокая ретикулоцитопения;
- нормоклеточный костный мозг с изолированным снижением содержания эритроидных предшественников;
- нормальное или незначительно сниженное число гранулоцитов;
- нормальное или незначительно повышенное число тромбоцитов.
Уровень фетального гемоглобина, хотя и может быть повышен, не служит диагностическим признаком. Редко у больных анемией Даймонда-Блекфена с первых месяцев жизни в костном мозге повышено количество примитивных эритробластов, которые могут быть приняты за лейкемические бласты, что приводит к ошибочной диагностике лейкоза. С возрастом клеточность костного мозга, определяемая по трепанобиопсии, может значительно снижаться, у некоторых больных развивается умеренная тромбоцитопения. Специализированные исследования позволяют выявить резко сниженное количество коммитированных предшественников эритропоэза — бурстобразующих единиц эритроцитов и колониеобразующих единиц эритроцитов. Уровень эритропоэтина у больных анемией Даймонда-Блекфена резко повышен.
Дифференцировать анемию Даймонда-Блекфена необходимо от других форм ПККА у детей, главным образом от ТЭД. Документация нормального уровня гемоглобина до клинического проявления анемии и самостоятельное разрешение синдрома свидетельствуют против анемии Даймонда-Блекфена.
[13], [14], [15], [16]
Единственной эффективной группой препаратов в лечении анемии Даймонда-Блекфена выступают глюкокортикостероиды. Обычно лечение начинают с преднизолона внутрь в дозе 2 мг/кг в сут. Ретикулоцитарный ответ ожидается через 2 нед, вслед за которым следует подъём уровня гемоглобина. После выхода значений гемоглобина на плато дозу преднизолона необходимо постепенно снижать до минимальной, позволяющей поддерживать уровень гемоглобина выше 90 г/л. Зачастую для поддержания гематологического ответа достаточно использовать дозы в районе 2,5-5 мг в сут или через день. Если на стандартные дозы преднизолона ответа не получено, оправдано применение повышенных доз — 5 мг/кг в сут. Повышенные дозы возможно применять в режиме пульс-терапии по 7 дней с последующим 2-недельным перерывом. Всего проводят 3-4 пульс-терапии. При достижении ответа можно либо увеличивать интервалы между курсами, либо переводить пациента на ежедневный приём глюкокортикостероидов в стандартных дозах с последующим снижением до минимально эффективных доз. Использование сверхвысоких доз метилпреднизолона — 30-100 мг/кг, несмотря на относительную популярность, не доказало своей высокой эффективности. В целом к применению глюкокортикостероидов чувствительно около 70% пациентов, однако 20% ответивших впоследствии становятся резистентными к ним. Интересно, что из больных, первично не ответивших на глюкокортикостероиды, часть отвечает при последующих попытках, поэтому пробное лечение глюкокортикостероидами необходимо время от времени возобновлять (1 раз в 1-2 года).
Лечение пациентов с анемией Даймонда-Блекфена ростовыми факторами — интерлейкином-3 и эритропоэтином, несмотря на лабораторные предпосылки, оказалось абсолютно неэффективным. Место циклоспорина, несмотря на несколько отдельных сообщений об успешном лечении, в терапии больных с анемией Даймонда-Блекфена сомнительно. Аллогенную трансплантацию костного мозга можно предложить больным, имеющим HLA-геноидентичного сиблинга, если они не чувствительны к лечению глюкокортикостероидами.
Больным, у которых глюкокортикостероиды неэффективны или эффективны в дозах, вызывающих неприемлемые долгосрочные побочные эффекты (остеопороз, нарушения роста, диабет, катаракту, синдром Кушинга), необходимо проведение грамотной трансфузионной и хронической хелационной терапии деферроксамином и/или деферипроном.
В литературе приводят данные катамнеза 200 детей с анемией Даймонда-Блекфена — у 22,5% возникла спонтанная ремиссия; у 41,8% — кортикостероидзависимая; у 35,7% — трансфузии — зависимая ремиссия; 27,6% детей умерли.
[17]
источник
Исключены: аутоиммунная болезнь (системная) БДУ (M35.9)
болезнь, вызванная вирусом иммунодефицита человека ВИЧ (B20 — B24)
врожденные аномалии (пороки развития), деформации и хромосомные нарушения (Q00 — Q99)
новообразования (C00 — D48)
осложнения беременности, родов и послеродового периода (O00 — O99)
отдельные состояния, возникающие в перинатальном периоде (P00 — P96)
симптомы, признаки и отклонения от нормы, выявленные при клинических и лабораторных исследованиях, не классифицированные в других рубриках (R00 — R99)
травмы, отравления и некоторые другие последствия воздействия внешних причин (S00 — T98)
эндокринные болезни, расстройства питания и нарушения обмена веществ (E00 — E90).
Примечание. Все новообразования (как функционально активные, так и неактивные) включены в класс II. Соответствующие коды в этом классе (например, Е05.8, Е07.0, Е16-Е31, Е34.-) при необходимости можно использовать в качестве дополнительных кодов для идентификации функционально активных новообразований и эктопической эндокринной ткани, а также гиперфункции и гипофункции эндокринных желез, связанных с новообразованиями и другими расстройствами, классифицированными в других рубриках.
Исключены:
отдельные состояния, возникающие в перинатальном периоде (P00 — P96),
некоторые инфекционные и паразитарные болезни (A00 — B99),
осложнения беременности, родов и послеродового периода (O00 — O99),
врождённые аномалии, деформации и хромосомные нарушения (Q00 — Q99),
болезни эндокринной системы, расстройства питания и нарушения обмена веществ (E00 — E90),
травмы, отравления и некоторые другие последствия воздействия внешних причин (S00 — T98),
новообразования (C00 — D48),
симптомы, признаки и отклонения от нормы, выявленные при клинических и лабораторных исследованиях, не классифицированные в других рубриках (R00 — R99).
Chapter IX Diseases of the circulatory system (I00-I99)
Исключено:
болезни эндокринной системы, расстройства питания и нарушения обмена веществ (E00-E90)
врожденные аномалии, деформации и хромосомные нарушения (Q00-Q99)
некоторые инфекционные и паразитарные болезни (A00-B99)
новообразования (C00-D48)
осложнения беременности, родов и послеродового периода (O00-O99)
отдельные состояния, возникающие в перинатальном периоде (P00-P96)
симптомы, признаки и отклонения от нормы, выявленные при клинических и лабораторных исследованиях, не классифицированные в других рубриках (R00-R99)
системные нарушения соединительной ткани (M30-M36)
травмы, отравления и некоторые другие последствия воздействия внешних причин (S00-T98)
транзиторные церебральные ишемические приступы и родственные синдромы (G45.-)
This chapter contains the following blocks:
I00-I02 Acute rheumatic fever
I05-I09 Chronic rheumatic heart diseases
I10-I15 Hypertensive diseases
I20-I25 Ischaemic heart diseases
I26-I28 Pulmonary heart disease and diseases of pulmonary circulation
I30-I52 Other forms of heart disease
I60-I69 Cerebrovascular diseases
I70-I79 Diseases of arteries, arterioles and capillaries
I80-I89 Diseases of veins, lymphatic vessels and lymph nodes, not elsewhere classified
I95-I99 Other and unspecified disorders of the circulatory system
источник
врожденная апластическая анемия
заместительные трансфузии эритроцитной массы
АДБ – анемия Даймонда — Блекфена
ГКС — глюкокортикоидная терапия
ДНК – дезоксирибонуклеиновая кислота
МДС – миелодистпластический синдром
МРТ – магниторезонансная томография
НЖСС – ненасыщенная железосвязывающая способность сыворотки
НТЖ – насыщение трасферина железом
ОЖСС — общая железосвязывающая способность сыворотки
ОМЛ – острый миелобластный лейкоз
ТГСК — трансплантация гемопоэтических стволовых клеток
УЗИ – ультразвуковое исследование
AB0 – группа крови по системе AB0
eADA – эритроцитарная аденозиндезоминаза
HLA – главный комплекс гистосовместимости
MCH – средняя концентрация гемоглобина
per os – внутрь (перевод с латинского)
p53 – внутриклеточный белок, который защищает организм от последствий повреждения ДНК, инициируя либо арест клеточного цикла, либо апоптоз поврежденных клеток, основной опухолевый суппресор
Агранулоцитоз – число нейтрофилов менее 0,5х109/л
Аллоиммунизация — выработка антител к антигенам других людей.
Анемия – снижение содержания гемоглобина.
Апоптоз – естественная гибель клеток.
Арест клеточного цикла – остановка деления клеток.
Гаплонедостаточность – недостаточность половинного количества генного продукта для нормального функционирования организма.
Гепатоспленомегалия – увеличение размеров печени и селезенки.
Гипертелоризм – широко поставленные глаза.
Готическое небо – высокое небо.
Вакцинопрофилактика – введение вакцин для профилактики инфекций.
Коарктация аорты – сужение аорты.
Лейкодеплетированная – очищенная от примесей лейкоцитов.
Микрогнатия – челюсть уменьшенного размера.
Микроотия – маленькие ушные раковины.
Небная расщелина — незаращение верхнего неба.
Нейтропения – снижение числа нейтрофилов в периферической крови.
Нормохромная анемия – анемия с нормальным цветовым показателем (нормальным MCH) эритроцитов.
Остеоденситометрия – измерение минеральной плотности костей.
Ретикулоцитопения – снижение числа ретикулоцитов в периферической крови.
Синдактелия – сращение пальцев.
Тромбоцитоз — повышение числа тромбоцитов в периферической крови.
Тромбоцитопения – снижение числа тромбоцитов в периферической крови.
Хелатор – вещество, образующее устойчивое нетоксичное соединение с металлом (в данном случае с железом), способное покинуть организм.
Хелаторная терапия – использование хелаторов с лечебной целью.
Эритробластопения – снижение числа эритробластов в костном мозге.
Анемия Даймонда – Блекфена — редкая форма врожденной аплазии кроветворения, в основном красноклеточной (эритроидной), раннего и детского возраста, развивающаяся в результате апоптоза эритроидных предшественников в костном мозге вследствие дефекта биосинтеза рибосом.
Синдром Пирсона – мультисистемное заболевание с преобладающим вовлечением кроветворения, поджелудочной жлезы и печени, развивающиеся вследствии дефекта митохондрильной ДНК.
Рибосомопатии — группа генетически детерминированных расстройств, являющихся результатом нарушения синтеза рибосом или их функциональных дефектов.
Анемия Даймонда-Блекфена — редкая форма врожденной аплазии кроветворения, в основном красноклеточной (эритроидной), раннего и детского возраста, развивающаяся в результате апоптоза эритроидных предшественников в костном мозге вследствие дефекта биосинтеза рибосом [1-5].
В настоящее время данные о генетических нарушениях (повреждение рибосом за счет нарушения формирования их субъединиц), лежащих в основе патогенеза АДБ, приводящих к гаплонедостаточности рибосом, позволяют отнести данное заболевание к группе рибосомопатий [5,6]. Мутации в генах рибосомальных белков приводят к нарушению синтеза как малых, так и больших субъединиц рибосом, что индуцирует р53 с последующим арестом клеточного цикла на границе фаз G1/S [7-12], было показано, что данные изменения не затрагивают другие клеточные линии [9]. При этом известно, что мутации в различных генах рибосомальных белков по-разному влияют на дифференцировку клеток эритроидного ряда [13,14]. Например, мутации в гене RPS19 индуцировали снижение пролиферации клеток-предшественников, однако конечная дифференцировка эритроцитов оставалась ненарушенной. В то же время мутации в гене RPL11 приводили не только к резкому подавлению пролиферации эритроидных предшественников, но и к торможению дифференцировки эритроцитов и значительному увеличению апоптоза в культуре клеток [15].
Еще одним объяснением нарушения эритропоэза может служить тот факт, что эритроидная дифференцировка сопровождается разительной перестройкой ядерных структур с конденсацией хроматина, являющейся подготовительным шагом для утраты ядра. Вследствие этого ядрышко подвергается структурным и молекулярным изменениям, что может потенцировать рибосомальный стресс, вызванный мутациями в рибосомальных белках, и вести к апоптозу [16]. В норме транскрипты будущих рибосомальных белков образуются в ядре РНК-полимеразой II, транслируются в цитоплазме, после чего данные белки транспортируются в ядрышко, где принимают участие в формировании рибосом. 40S и 60S субъединицы рибосом затем эспортируются из ядрышка через нуклеоплазму в цитоплазму, где соединяются в 80S субъединицей рибосомы и выполняют свою роль в синтезе белка в клетке [17].
По некоторым данным, мутации в генах RPL5 и RPL11 чаще ассоциируются с наличием врожденных аномалий, чем мутации в гене RPS19, причем первые характеризуются более тяжелым фенотипом [18].
В настоящее описан большой спектр мутаций и делеций различных генов рибосомальных белков, наиболее часто встречаемые поломки в генах RPS19, RPS10, RPS24, RPS26, RPL5, RPL11, RPL35a, RPS7, RPS17 [19-21]
Идентифицированы также единичные случаи АДБ в результате мутации генов GATA1, FLVCR1 и TFR2 [22,23].
По данным Kynaston et al (1993) расчетная частота встречаемости заболевания составляет 1 на 100000 или 1 на 200000 рожденных живыми детей [24], по данным других авторов – 5-7 на 1000000 рожденных живыми детей вне зависимости от национальности и пола [25-27]. По данным Российского регистра в ежегодно в стране рождается 8-11 детей с АДБ, что в среднем составляет 4,975 случаев на 1000000 рожденных живыми детей. Около 45% больных – семейные случаи с аутосомно-доминантным путем наследования, оставшиеся 55% больных – спорадические случаи [27].
Кумулятивный риск развития всех злокачественных новообразований у больных АБД превышает общепопуляционный в 5,4 раза. Максимальный риск развития был отмечен для миелодиспластического синдрома, острого миелобластного лейкоза, аденокарциномы толстой кишки, остеогенной саркомы и злокачественных опухолей женских половых органов [9].
Не менее 40% пациентов с АДБ нуждаются в проведении постоянной трансфузионной терапии [2]. Около 75% пациентов с АДБ доживают до возраста 40 лет, для трансфузионно-зависимых пациентов этот показатель составляет чуть более 57% [10].
D61.0 – конституциональная апластическая анемия
Общепринятой классификации анемии Даймонда-Блекфена не существует, однако для детализации состояния заболевания эксперты предлагают выделять:
трансфузионно зависимую (пациент получает регулярные заместительные трансфузии эритроцитной массы)
полную медикаментозную компенсацию (у пациента достигнут полный гематологический ответ на терапию ГКС)
медикаментозная субкомпенсация (у пациента достигнут частичный гематологический ответ на терапию ГКС)
спонтанная компенсация (у пациента произошла спонтанная полная гематологическая компенсация)
Основная жалоба – бледность кожи и слизистых, слабостью, утомляемостью (у детей первых месяцев жизни проявляется быстрым утомлением при кормлении, особенно грудью матери) [1-3,5]. В дальнейшем (у детей старше 1 года) присоединяются жалобы на отставание физического роста ребенка [1-3,5].
Сбор анамнеза при АДБ подразумевает тщательный расспрос о возрасте появления первых симптомов заболевания, наличие в семье детей или взрослых с аналогичными проявлениями (заболеванием) [1-3,5].
Средний возраст начала клинических проявлений – 2 месяца жизни, средний возраст установления диагноза – 3-4 месяца [1-3,5]. В более 90% случаев манифестация заболевания на первом году жизни, крайне редко – в первые сутки жизни [1-3,5].
Общий осмотр подразумевает оценку общего физического состояния, роста и массы тела, наличия вторичных половых признаков в соответствующем возрасте., выявление врожденных аномалий развития. Более чем у половины больных АДБ выявляются врожденные аномалии развития. Пороки развития, кроме низкого роста, встречаются в 47% случаев: аномалии черепа и лицевого скелета (гипертелоризм, высокий выпуклый лоб, готическое небо, небная расщелина, плоская спинка носа, микрогнатия, микроцефалия, микротия, низко расположенные ушные раковины) – 50%, и аномалии кистей рук (удвоенный, расщепленный, 3-фаланговый большой палец, синдактилия) – 38%, патология сердца (дефект межжелудочковой перегородки, дефект межпредсердной перегородки, коарктация аорты, тетрада Фалло) – 30%, и мочеполовой системы (подковообразная почка, удвоение мочевыводящих путей, гипоспадия – 39%, сочетанные пороки развития встречаются в 21% случаев [1-5,21,24-29].
Физическое развитие низкое. Низкий вес при рождении встречается в 10% случаев, при этом в половине из этих случаев отмечается отставание физического развития от гестационного возраста. Более 60% больных имеют рост менее 25 перцентиля [1-5,21,24-29].
Оценивая причину низкого роста у пациентов АДБ трудно отделить конституциональные особенности от побочных эффектов проводимой терапии (перегрузка железом вследствие постоянных гемотрансфузий или длительный прием глюкокортикостероидов) [1,2,5,28-30].
- Диагноз АДБ рекомендовано устанавливать на основании клинических проявлений и данных лабораторного обследования [1-5,24-28].
- • Нормохромная, обычно макроцитарная, анемия в раннем возрасте без вовлечения других клеточных линий.
- • Ретикулоцитопения.
- • Нормоклеточный костный мозг с селективным уменьшением эритроидных предшественников ( 100 г/л, нормальное число ретикулоцитов;
частичный — Hb 85-100 г/л, наличие ретикулоцитов;
отсутствие ответа — Hb 500 мкг/л (уровень убедительности доказательства В) [1-5,28,30,34-47]. Отменяться хелаторная терапия может при достижении верхней границы возрастной нормы содержания ферритина сыворотки при условии прекращения заместительной трансфузионной терапии и нормализации содержания железа в печени и миокарде, оцененных методом МРТ Т2* (для печени возможно методом определения содержания железа в сухом веществе печени). Хелаторы: деферазирокс (начальная доза 30 м г/кг/сут peros ежедневно, далее с шагом 5 м г/кг/сут повышается до максимальной дозы 45 м г/кг/сут или понижается в зависимости от концентрации ферритина сыворотки) (уровень убедительности доказательства В) [1-5,27,28,30,35-38,45,46], при содержании ферритина сыворотки менее 500 мкг/л доза снижается до 125-250 мг/сут (уровень убедительности доказательства С) [30,37,45,46], дефероксамин (начальная доза 40 мг/кг/сут подкожно 5 дней в неделю в виде длительной инфузии (8-12 ч), при необходимости интенсивной хелации, в случае развития застойной сердечной недостаточности 100 мг/кг/сут непрерывно внутривенно капельно в течение 7-10 дней (уровень убедительности доказательства В) [28,30,45-47]). Для интенсификации хелаторной терапии может использоваться комбинация деферазирокса (30 мг/кг/сут per os ежедневно) в сочетании с дефероксамином (40-50 мг/кг/сут подкожно ежедневно в течение 8-12 ч) (уровень убедительности доказательства B) [28,30,38-47]. Применение деферипрона в качестве препарата 1-й линии нецелесообразно в связи с высоким риском развития агранулоцитоза, и его назначение возможно только при наличии противопоказаний к деферазироксу и дефероксамину.
При проведении хелаторной терапии необходимо контролировать:
сывороточное железо, ОЖСС/НЖСС, НТЖ, сывороточный ферритин каждые 3 мес при подборе дозы хелатора, далее каждые 6 мес;
клиренс эндогенного креатинина до начала хелаторной терапии, каждые 3 мес на этапе подбора дозы, далее каждые 6-12 мес;
МРТ Т2* печени и миокарда 1 раз в год.
3.3 Хирургическое лечение
При данной патологии не используется.
3.4 Иное лечение
Трансплантация гемопоэтических стволовых клеток рассматривается в качестве радикальной терапии [1,2,4,5,27,28,30,48].
- При отсутствии эффекта на ГКС-терапию трансплантация гемопоэтических стволовых клеток (ТГСК) от родственного или неродственного HLA-совместимого донора рекомендуется рассматривать как альтернатива пожизненной заместительной терапии эритроцитной массой для пациентов младше 9 лет [1,4,5,27,28,30,48].
Уровень убедительности доказательства В
- ТГСК рекомендуется рассматривать как радикальный метод лечения для пациентов младше 9 лет в случае наличия родственного HLA-совместимого донора у трансфузионно зависимых пациентов, не отвечающих на глюкокортикоиды и учитывая риск прогрессивного угнетения кроветворения и развития злокачественных заболеваний у больных АДБ, ответивших на ГКС-терапию [1,2,4,5,27,28,30,48].
уровень убедительности доказательства В
Комментарий: В настоящее время, по данным регистров АДБ Франции и Германии, бессобытийная выживаемость при проведении ТГСК от родственного HLA-совместимого донора в возрасте младше 9 лет составляет 94%, а в более старшем возрасте 55%. При этом родственный донор должен быть обследован для исключения субклинической формы АДБ [1,2,4,5,27,28,30,48].
- У пациентов младше 9 лет неродственную HLA-совестимую ТГСК рекомендовано рассматривать как вариант радикальной терапии [1,2,27,28,48]
Уровень убедительности доказательства С .
Комментарий: В настоящее время, по данным регистров АДБ Франции и Германии, бессобытийная выживаемость при проведении ТГСК от неродственного HLA-совместимого донора пациента младше 9 лет составляет 85% [27,28,48].
Уровень убедительности доказательства D
Комментарий: Описаны случаи достижения ремиссии при использовании лейцина (по 800 мг/кг/м2 три раза в сутки), однако в клинических рандомизированных исследованиях эффективность такой терапии не доказана [1,2,5,30,49-51].глюкокортикостероидная терапия
Специфических реабилитационных мероприятий в отношении пациентов с АДБ не разработано. Пациенты с АДБ вне зависимости от возраста и получаемой терапии могут посещать детские дошкольные, школьные учреждения, пребывать в оздоровительных лагерях, заниматься в физической культурой и спортом (бесконтактные виды спорта).
После установления диагноза, выбора лечебной тактики, подбора доз препаратов пациент передается под диспансерное наблюдение педиатра (если есть должность, то под наблюдение гематолога) по месту жительства. Терапия проводится амбулаторно, длительно/пожизненно. Больные и члены их семей должны быть подробно ознакомлены как с сутью заболевания, возможным осложнениям проводимой терапии, так и обучены правилам индивидуальной гигиены.
Профилактическая вакцинация проводиться в соответствии с Национальным календарем. Необходимые мероприятия при диспансерном наблюдении представлены в таблице 1.
Заместительная терапия эритроцитной массой без/с хелаторной терапией
Начало терапии – каждые 2 недели до завершения подбора дозы
Последующее наблюдение – 1 раз в 3 месяца
осмотр прозрачных сред глаза с медикаментозным расширением зрачка – 1 раз в 12 мес.
1 раз в год, при выявлении патологии чаще
Для пациентов старше 5 лет – 1 раз в год
Холтеровское мониторирование сердечного ритма
Для пациентов старше 2 лет в случае сохранения ферритина сыворотки более 1000 мкг/л в двух последовательных анализах — 1 раз в год
Мониторирование суточного артериального давления
Для пациентов старше 2 лет в случае сохранения ферритина сыворотки более 1000 мкг/л в двух последовательных анализах — 1 раз в год
МРТ в режиме Т2* печени, миокарда, поджелудочной железы и гипофиза
Для пациентов 5-ти лет и старше 1 раз в год
УЗИ органов брюшной полости и почек
ARFI-эластография печени, поджелудочной железы
Для пациентов старше 2 лет – 1 раз в год
Общий анализ крови с подсчетом ретикулоцитов, тромбоцитов, лейкоцитарной формулы
каждые 2 недели до завершения подбора дозы, далее 1 раз в 3 месяца
Биохимический анализ крови (мочевина, креатинин, общий билирубин, прямой билирубин, АЛТ, АСТ, ЛДГ, ЩФ, глюкоза, К + , Na + )
1 раз в 1 месяц при подборе дозы хелатора, далее 1 раз в 3 месяца
Клиренс эндогенного креатинина
Каждые 6-12 месяцев при проведении хелаторной терапии
Сывороточное железо, ОЖСС (НЖСС), НТЖ, ферритин сыворотки
До начала хелаторной терапии 1 раз в 6-12 месяцев; при подборе дозы хелатора каждые 3 месяца, далее каждые 6 месяцев
Антиэритроцитарные антитела (непрямая и прямая пробы Кумбса)
Перед каждой трансфузией эритроцитной массы
иммунофенотипирование эритроцитов по системе АВ0, Rh-фактору и редким группам крови (Kell и др.)
содержание витамина D в сыворотке крови
Для пациентов старше 2 лет – 1 раз в год
Т4 свободный (тироксин свободный)
IGF-I, инсулиноподобный фактор роста
Для пациентов старше 7 лет – 1 раз в год
Для пациентов старше 2 лет – 1 раз в год
B-Cross Laps в сыворотке крови
Кальций общий в сыворотке крови
Кальций ионизированный в сыворотке крови
Для пациентов старше 7 лет – 1 раз в год
В целом прогноз для жизни достаточно благоприятный. Выполнение данных клинических рекомендаций позволяет сохранить полноценную работоспособность пациента. Продолжительность жизни ограничена в первую очередь развитием осложнений от проводимой терапии.
Спонтанная ремиссия АДБ возможна в примерно 20% случаев к 25 годам независимо от ранее проводимой терапии [1,2,5,24,25,27,30].
Осложнение заместительной терапии эритроцитной массой – посттрансфузионная перегрузка железом – может существенно сокращать продолжительность жизни и ухудшать качество жизни больных [1,2,5,24,25,27,30,34].
Продолжительность жизни больных: до 40 лет доживает 75,1±4,8% больных; в случае достижения ремиссии или медикаментозной ремиссии выживаемость составляет 85-100%; трансфузионно зависимые пациенты доживают до взрослого возраста в 60% случаев [1,4,24,25,27].
Общая выживаемость после родственной совместимой ТГСК, если она проводилась до 9-летнего возраста, составляет 95%, после неродственной полностью совместимой ТГСК – 85% [1,2,4,5,30,48].
Смертность пациентов с АДБ зависит от развития и степени тяжести осложнений от проводимой терапии (посттрансфузионная перегрузка железом, инфекции, осложнения после ТГСК) – 67%, связана с прогрессией заболевания (тяжелая аплазия кроветворения, злокачественные заболевания) – 22%, не установлена причинная связь – 11% случаев [3,24,25,27].
При отсутствии ТГСК избегать профессий, связанных возможными травмами. При успешной ТГСК ограничений в выборе профессии нет.
Без ТГСК детородная функция обычно не страдает, после проведенной ТГСК возможно бесплодие.
Физиологические изменения, происходящие при беременности, могут вызвать повышение потребности как в ГКС, так и в трансфузиях эритроцитной массы. Во время беременности может быть возврат клинических проявлений и необходимость терапии у больных со спонтанной компенсацией АДБ.
Пренатальная диагностика и генетическое консультирование:
При генетическом консультировании необходимо учитывать высокую вероятность рождения больного ребенка в данной семье при последующих беременностях. Пренатальная диагностика возможна при идентифицированной мутации у пациента; в
30% случаев мутацию выявить не удается, следовательно проведение пренатальной диагностики становиться невозможно.
При генетическом консультировании необходимо учитывать крайне высокую вероятность рождения больного ребенка.
У пациентов мужского пола при идентифицированной мутации возможно проведение генетического исследования спермы для определения риска передачи данного заболевания следующему поколению. В случае выявления только мутантного аллеля – риск рождения больного ребенка составляет 100%, деторождение не рекомендуется. В случае наличия нормального и мутантного аллеля – показана ЭКО с предимплантационной диагностикой.
У пациентов женского пола при идентифицированной мутации возможно ЭКО с предимплантационной диагностикой.
Критерий качества
На этапе первичной диагностики выполнен общий анализ крови с подсчетом ретикулоцитов и тромбоцитов
На этапе первичной диагностики выполнено морфологическое исследование пунктата костного мозга
На этапе первичной диагностики выполнено исследование содержания лактата в венозной крови
На этапе первичной диагностики выполнено исследование фракций гемоглобина
На этапе первичной диагностики выполнен тест на ломкость хромосом (диэпоксибутановый тест или тест с митомицином С)
На этапе первичной диагностики выполнено стандартное кариотипирование
На этапе первичной диагностики выполнено исследование параметров обмена железа (железо сыворотки, НЖСС или ОЖСС, НТЖ, ферритин сыворотки)
Выполнено в течении первых 30 дней после установки диагноза HLA-типирование сиблингов (при их наличие)
Выполнено в течении 60 дней после установки диагноза ДНК-исследование генов рибосомальных белков
Выполнены обследования, направленные на контроль эффективности и безопасности лечения (осмотр прозрачных сред глаза, УЗИ органов брюшной полости и почек, ЭХО-КГ, антропометрия, исследование ферритина сыворотки, исследование содержания железа в печени и миокарде методом МРТ Т2*) не реже 1 раза в год
Проведено фенотипирование эритроцитарных антигенов по системе AB0, Rh, Kell перед трансфузией эритроцитной массы не реже 1 раза в сутки (при проведении трансфузионной терапии эритроцитарной массой)
Выполнен индивидуальный подбор эритроцитной массы для трансфузии не реже 1 раза в сутки (при проведении трансфузионной терапии эритроцитарной массой)
Выполнена хелаторная терапия в случае повышения ферритина сыворотки более 600 мкг/л при регулярных трансфузиях эритроцитной массы не менее 30 суток отмомента выявления
Проведена хелаторная терапия ежедневно постоянно в случае выявленной перегрузки железом
Проведена терапия глюкокортикостероидами начата в возрасте 8 месяцев и старше*
* — использование глюкокортикостероидов младше 8 месяцев категорически запрещено в связи с развитием необратимых тяжелых осложнений;
** — использование большей чем 0,5 мг/кг/сут дозы глюкокортикостероидов для поддержания гемоглобина более 90 г/л – запрещено в связи с развитием необратимых осложнений; если требуется большая доза для поддержания гемоглобина 90 г/л и выше – терапия глюкокортикостероидами должна быть признана не эффективной и прекращена; использование глюкокортикостероидов совместно с переливаниями эритроцитной массы – запрещено.
Vlachos A, Ball S, Dahl N, Alter BP, Sheth S, Ramenghi U, et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol. 2008; 142(6):859-76.
Ball S. Diamond Blackfan anemia. American Society of Hematology Education Program 2011; 2011: 487-91.
Orfali KA, Ohene-Abuakwa Y, Ball SE. Diamond Blackfan anaemia in the UK: clinical and genetic heterogeneuty. Br J Haematol. 2004;125(2): 243-52.
Lipton JM, Atsidaftos E, Zyskind I, Vlachos A. Improving clinical care and elucidating the pathophysiology of Diamond Blackfan anemia: An update from the Diamond–Blackfan Anemia Registry. Pediatr. Blood & Cancer. 2006; 46: 558–64.
Jaako P, Flygare J, Karisson S. Diamond-Blackfan anemia: pathogenesis, management and development of future therapies. Hematology education: the education program for the annual congress of the European Hematology Association. 2013;7:101-8
Narla A, Ebert BL Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010;115(16):3196-205
Fumagalli S, Thomas G. The role of p53 in ribosomopathies. Semin Hematol. 2011;48(2):97-105.
Zhang Y, Lu H. Signaling to p53: ribosomal proteins find their way. Cancer Cell. 2009; 16(5):369-77
Dutt S, Narla A, Lin K, Mullally A, Abayasekara N, Megerdichian C, et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood. 2011;117(9):2567-76
Horn HF, Vousden KH. Cooperation between the ribosomal proteins L5 and L11 in the p53 pathway. Oncogene. 2008;27(44):5774-84
Chen D, Zhang Z, Li M, Wang W, Li Y, Rayburn ER, et al. Ribosomal protein S7 as a novel modulator of p53-MDM2 interaction: binding to MDM2, stabilization of p53 protein, and activation of p53 function. Oncogene. 2007;26(35):5029-37.
Fumagalli S, Ivanenkov VV, Teng T, Thomas G. Suprainduction of p53 by disruption of 40S and 60S ribosome biogenesis leads to the activation of a novel G2/M checkpoint. Genes Dev. 2012;26(10):1028-40
Gazda HT, Kho AT, Sanoudou D, Zaucha JM, Kohane IS, Sieff CA, et al. Defective ribosomal protein gene expression alters transcription, translation, apoptosis, and oncogenic pathways in Diamond-Blackfan anemia. Stem Cells. 2006;24(9):2034-44
Badhai J, Fr?jmark AS, J Davey E, Schuster J, Dahl N. Ribosomal protein S19 and S24 insufficiency causes distinct cell cycle defects in Diamond-Blackfan anemia. Biochim Biophys Acta. 2009;1792(10):1036-42
Moniz H, Gastou M, Leblanc T., Hurtaud C., Cr?tien A. L?cluse Y., et al. Primary hematopoietic cells from DBA patients with mutations in RPL11 and RPS19 genes exhibit distinct erythroid phenotype in vitro. Cell Death Dis. 2012;3:e356
Horos R, Ijspeert H, Pospisilova D, Sendtner R., Andrieu-Soler C, Taskesen E, et al. Ribosomal deficiencies in Diamond-Blackfan anemia impair translation of transcripts essential for differentiation of murine and human erythroblasts. Blood. 2012; 119(1): 262-72
Horos R, von Lindern M. Molecular mechanisms of pathology and treatment in Diamond Blackfan anaemia. Br J Haematol. 2012;159(5):514-27
Gazda HT, Sheen MR, Vlachos A, Choesmel V, O»Donohue MF, Schneider H, et al. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am J Hum Genet. 2008;83(6):769-80
Boria I, Garelli E, Gazda HT, Aspesi A, Quarello P, Pavesi E, Ferrante D, Meerpohl JJ, Kartal M, Da Costa L, Proust A, Leblanc T, Simansour M, Dahl N, Fr?jmark AS, Pospisilova D, Cmejla R, Beggs AH, Sheen MR, Landowski M, Buros CM, Clinton CM, Dobson LJ, Vlachos A, Atsidaftos E, Lipton JM, Ellis SR, Ramenghi U, Dianzani I. The ribosomal basis of Diamond–Blackfan anemia: mutation and database update. Hum Mutat. 2010: 31:1269–1279
Vlachos A, Dahl N, Dianzani I, Lipton JM: Clinical utility gene card for: Diamond Blackfan anemia-update 2013. Euro J Hum Genet, doi: 10.1038/jejhg.2013.34
Smetanina NS, Mersiyanova IV, Kurnikova MA, Ovsyannikova GS, Hachatryan LA, Bobrynina VO, Maschan MA, Novichkova GA, Lipton JM, Maschan AA. Clinical and Genomic Heterogeneity of Diamond Blackfan Anemia in the Russian Federation. Pediat Blood & Cancer, 2015; 62(9): 1597-1600
Sankaran VG, Ghazvinian R, Do R, Thiru P, Vergilio JA, Beggs AH, et al. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J Clin Invest. 2012; 122(7):2439-43
Rey MA, Duffy SP, Brown JK, Kennedy JA, Dick JE, Dror Y, et al. Enhanced alternative splicing of the FLVCR1 gene in Diamond Blackfan anemia disrupts FLVCR1 expression and function that are critical for erythropoiesis. Haematologica. 2008; 93(11):1617-26
Kynaston J.A., West N.C., Reid M.M. A regional experience of red cell aplasia. Eur J Pediatr. 1993; 152: 306-308
Ball S.E., McGuckin C.P., Jenkins G., Gordon-Smith E.C. Diamond-Blackfan anaemia in the U.K.: Analysis of 80 cases from a 20-year birth cohort. Br J Haematol. 1996;94:645–653
Campagnoli M.F., Garelli E., Quarello P., Carando A., Varotto S., Nobili B., Longoni D., Pecile V., Zecca M., Dufour C. Molecular basis of Diamond-Blackfan anemia: New findings from the Italian registry and a review of the literature. Haematologica. 2004;89:480–9
Willig T.N., Niemeyer C.M., Leblanc T., Tiemann C., Robert A., Budde J., Lambiliotte A., Kohne E., Souillet G., Eber S. Identification of new prognosis factors from the clinical and epidemiologic analysis of a registry of 229 Diamond-Blackfan anemia patients. DBA group of Soci?t? d»H?matologie et d»Immunologie P?diatrique (SHIP), Gesellshaft f?r P?diatrische Onkologie und H?matologie (GPOH), and the European Society for Pediatric Hematology and Immunology (ESPHI) Pediatr. Res.1999;46:553–61
Lipton JM, Ellis SR. Diamond-Blackfan anemia: diagnosis, treatment, and molecular pathogenesis. Hematol Oncol Clin North Am. 2009;23(2):261-82
Pospisilova D, Cmejlova J, Ludikova B, Stary J, Cerna Z, Hak J, et al. The Czech National Diamond-Blackfan Anemia Registry: clinical data and ribosomal protein mutations update. Blood Cells Mol Dis. 2012; 48(4): 209-18.
Vlachos A, Muir E. How I treat Diamond-Blackfan anemia. Blood. 2010;116(19):3715-23
St Pierre TG, Clark PR, Chua-Anusorn W, Fleming AJ, Jeffrey GP, Olynyk JK, Pootrakul P, Robins E, Lindeman R. Noninvasive measurement and imaging of liver iron concentration using proton magnetic resonance. Blood. 2005; 105 (2):855–61
Juliano JL, Siqueira MHA, Nobrega de Oliveira KT,Avila LF, Gottlieb I, Lopes MU, Fernandes AM, Strecker R, Greiser A. Use of an accelerated protocol for rapid analysis of iron overload in the heart and liver: the All Iron Detected (AID) multicenter study. Journal of Cardiovascular Magnetic Resonance (JCMR). 2015; 17 (Suppl. 1): 062 jcmronline.com/content/pdf/1532-429X-17-S1-O62.pdf
Wood JC. Impact of iron assessment by MRI. In: Hematology 2011. American society of hematology Education book, 2011:443–50
Овсянникова Г.С., Терещенко Т.В., Ибрагимова Д.И., Новичкова Г.А., Митрофанова А.М., Сметанина Н.С. Комплексная оценка перегрузки железом у детей с трансфузионно-зависимыми врожденными анемиями. Педиатрия. 2016; 95(4): 42-9
Cappellini MD, Bejaoui M, Agaoglu L, et al. Iron chelation with deferasirox in adult and pediatric patients with thalassemia major: efficacy and safety during 5 years’ follow-up. Blood. 2011;118(4):884-893
Cappellini MD, Porter J, El-Beshlawy A, et al. Tailoring iron chelation by iron intake and serum ferritin: the prospective EPIC study of deferasirox in 1744 patients with transfusion-dependent anemias. Haematologica. 2010;95(4):557-566
Porter JB, Piga A, Cohen A, et al. Safety of deferasirox (Exjade) in patients with transfusion dependent anemias and iron overload who achieve serum ferritin levels 6 рожденных живыми новорожденных)
Острая (вирусная или идиопатическая)
Возраст к моменту постановки диагноза
6 мес. – 4 года (иногда старше)
90% к 1 году, из них 25% при рождении или в первые 2 мес.
источник
Протекает с угнетением всех ростков кроветворения (т.е. с общим поражением гемопоэза) и врожденными аномалиями развития. Наследуется по АР типу, встречаются семейные формы заболевания – у братьев и сестер.
Патогенез. Поломка дистального плеча одной из хромосом 1-3 пары. При анализе метафазных пластинок выявляются хроматидные и хромосомные разрывы – одиночные и парные фрагменты, «кресты». Предположительный механизм – повреждение ДНК свободными радикалами кислорода в результате дефекта систем антиоксидантной защиты (супероксиддисмутазы и каталазы).
Клиника. Заболевание чаще диагностируется в возрасте 4-5 лет, когда проявляется гематологическая симптоматика (реже – при рождении).
Задержка внутриутробного развития, у 70-90% больных — врожденные аномалии развития (микроцефалия, микроофтальмия, косоглазие, эпикант, недоразвитие большого пальца кисти, лучевой кости, синдактилия, аномалия развития ребер, аномалии почек, снижение слуха. Характерна бронзово-коричневая пигментация кожи за счет отложения меланина в клетках базального слоя эпидермиса + пятна «кофе с молоком», трофические нарушения кожи, ногтей и зубов.
Кровь. Панцитопения. Анемия нормохромная, анизоцитоз с тенденцией к макроцитозу, пойкилоцитоз. Ретикулоциты 2-2,5%, по мере прогрессирования заболевания их количество снижается. В эритроцитах – высокий уровень Фетального гемоглобина. Анемия сочетается с повышенным уровнем эритропоэтина в сыворотке крови. Стойкая лейкопения (до 0,1 Г/л), тромбоцитопения (до единичных тромбоцитов в мазке).
Костный мозг. Стренальный пунктат – гипоклеточный. На ранних стадиях базофильный КМ, вследствие задержки созревания эритрокариоцитов на стадии базофильных нормобластов, мегалобласты. Сужение гранулоцитарного ростка, задержка созревания гранулоцитов на стадии миелоцитов и метамиелоцитов. Снижение количества мегакариоцитов. По мере прогрессирования болезни – угнетение гемопоэза, разрастание жировой ткани.
Течение заболевания характеризуется чередованием периодов обострения и ремиссии. Без лечения через 2 года после диагностики панцитопении умирают 80% больных, через 4 года – 100%. У больных с анемией Фанкони имеется высокий риск трансформации заболевания в острый лейкоз или злокачественные опухоли другой системной локализации (например, ЖКТ).
Является тотальной формой наследственной апластической анемии. Наследуется АР. Протекает с панцитпенией, не сопровождается врожденными пороками развития. Гематологические нарушения отмечаются в раннем детском возрасте. Прогноз неблагоприятный.
Протекает с избирательным поражением эритропоэза.
Встречается во всех этнических группах. Особенно распространена во Франции, Скандинавии. 75% — спорадические случаи, возможно АД, АР и Х-сцепленное носительство.
Патогенез. Спорадический или наследственный внутриклеточный дефект механизмов сигнальной трансдукции или факторов транскрипции на этапе раннего гемопоэза (ПСКК, КОЕ-ГЭММ) (наследственный – вследствие мутации гена на хромосоме 19, кодирующего рибосомальный протеин S19) увеличение чувствительности клеток к апоптозу.
Клиника. Заболевание диагностируется в течение 1-го года жизни. Дети, как правило, рождаются недоношенными, с бледной кожей и другими признаками гипоксии – вялость или возбуждение, беспокойство, сонливость, отказ от еды, диспепсические расстройства. У 25% детей – врожденные пороки развития. Гепато- и спленомегалия. Некоторые больные имеют характерный фенотип: волосы цвета пакли, курносый нос, большая верхняя губа. У 100% больных в моче обнаруживается антраниловая кислота – продукт триптофана.
Кровь. Нормохромная макроцитарная анемия, ретикулоцитов мало (0-0,1%). Высокий уровень фетального гемоглобина. Количество лейкоцитов и тромбоцитов в первые годы – нормальное, затем умеренная тромбоцитопения и нейтропения из-за снижения клональной эффективности кроветворных предшественников.
Костный мозг. Увеличение гипоклеточности по мере прогрессирования болезни. Количество эритрокариоцитов – менее 5%. Миелоидный, мегакариоцитарный и лимфоидный ростки – не изменены.
Протекает обычно хронически со спонтанными ремиссиями. Основная причина смерти – гемосидероз. Известный педиатр Уиллоуби писал: «Постоянная гипоксия, нарушение утилизации железа, трансфузии эритроцитарной массы неуклонно ведут к гемосидерозу, который в дальнейшем становится «убийцей» больного ребенка». Возможна трансформация заболевания в острый лейкоз, солидные опухоли (гепато- или остеосаркому), лимфогранулематоз.
источник
Железодефицитная анемия (малокровие) обычно рассматривают скорее в качестве симптома другого заболевания или в виде состояния, возникающего при недостаточной концентрации в организме железа.
Причины и факторы, способствующие развитию анемического синдрома:
- Развивается болезнь у маленьких детей и взрослых, соблюдающих строгую диету, если они не получают необходимого количества железа с едой.
- На фоне нарушений в пищеварительной системе. Известно, что всасывание микроэлементов происходит в тонкой кишке (верхние отделы) и в желудке. А тем более, после удаления части желудка, способность пищеварительной системы к абсорбции нарушается.
- Анемический синдром может развиться из-за больших потерь крови. Это является основной причиной железодефицитных состояний. Часто синдром возникает у женщин из-за больших потерь крови во время обильной менструации и у тех, кто страдает от язвы пищеварительного тракта, рака желудка, геморроя, новообразований в толстой кишке.
- Сокращение времени жизни эритроцитов в крови или быстрое их разрушение. В норме срок жизни эритроцита – 4 месяца. В некоторых случаях причиной гемолиза является патология селезенки. Развивается гемолитическая или серповидно-клеточная анемия. При таком заболевании организмом вырабатывается аномальный гемоглобин.
При подозрении на анемию, важно оперативно обратится за помощью к специалисту. Врач поможет выявить причины и способы лечения болезни. Синдром приводит к снижению иммунитета, к упадку сил, ограничению работоспособности. Также анемия может стать важным сигналом о развитии других патологий и серьезных проблем в работе отдельных органов и их систем.
Диагноз ставят по анализу крови, а лечение, как правило, заключается в восстановлении количества железа с помощью лек. препаратов. Лекарства используют внутрь или вводят с помощью инъекций.
| Пол, возраст | Порог Нb (Г/Л) | Порог Нb (Г/%) |
| Дети от 3 месяцев до 5 лет | 110 | 11,0 |
| Дети от 5 до 12 лет | 115 | 11,5 |
| Дети от 12 до 15 лет | 120 | 12,0 |
| Мужчины от 15 лет | 130 — 160 | 13,0 — 16,0 |
| Женщины от 15 лет | 120 — 140 | 12,0 — 14,0 |
| Беременные женщины | 110 | 11,0 |
Можно выделить 3 основных механизма развития анемии:
- Вследствие нарушения процесса образования эритроцитов и образования гемоглобина. Такого рода механизм можно проследить при недостаточном поступлении железа, фолиевой кислоты и витамина В12, при заболеваниях костного мозга. Иногда дефицитное состояние может возникнуть из-за приема сверхбольших доз витамина С. При приеме сверх доз аскорбиновой к-ты происходит блокировка витамина В12 и нарушение процессов кроветворения.
- Острая потеря эритроцитов. Обычно – это последствия острых кровотечений, операций и травм. При хронических кровотечениях в небольших объемах причиной анемии выступает не просто потеря эритроцитов, но и недостаток железа на фоне хронической утраты крови.
- Анемический синдром — последствия от ускоренного разрушения эритроцитов крови. При нормальной работе костного мозга и иных органов кроветворения эритроциты живут порядка 4 месяцев, а затем разрушаются. При гемолитической анемии, гемоглобинопатии и так далее скорость разрушения эритроцитов выше, чем их производства. Иногда процесс протекает под влиянием внешних стимулов, химических веществ (уксус).
Рассмотрим различные виды у взрослых. Анемия является симптомом, а не отдельным заболеванием и может возникать при ряде болезней. Они, как правило, связаны с первичным поражением кроветворной системы или не зависят от нее. Невозможно провести четкую нозологическую классификацию анемий. Для этого используют принцип практической целесообразности. И в результате заболевание делят по единому классификационному признаку – по цветовому показателю.
Падение уровня гемоглобина в крови чаще всего наблюдают при одновременном снижении количества и качества эритроцитов. Любой вид анемии непременно приводит к снижению эффективности дыхательной функции крови, возникновению кислородного голодания тканей. Обычно это видно по бледности кожных покровов, слабости, высокой утомляемости, головным болям, головокружении, одышке, учащенному ритму сердца и т.д.
Врач назначает рутинное исследование мазка крови, в процессе анализа морфолог обязан указать на отклонения размера эритроцитов. Уменьшение может быть, как в меньшую (микроцитарная анемия), так и большую сторону (макроцитарная). Однако, если необходимую оценку производят без специальных микрометров, то она является весьма субъективной.
Анемии, которые могут развиться при болезнях почек, гипопластические, АХЗ и острую постгеморрагическую относят к нормоцитарным. Макроцитарные анемии бывают нормо- и гиперхромными, а микроцитарные – гипохромными.
Намного информативнее автоматический анализ крови, при котором соблюдается четкая стандартизация важных показателей. Учитывают средний корпускулярный объем (СКО), который можно измерить в фемтолитрах (fl, фл). Нормальное значение СКО составляет 80-90 фл и называется нормоцитозом. Если показатель повышен до 95 и более – то фиксируют развитие макроцитоза. При снижении менее 80 фл ставят диагноз микроцитоз. Есть у автоматического метода и свои недостатки: достаточно дорогое и чуткое оборудование, которому необходимо соответствующее обслуживание.
При замене цветового показателя на СКО не нарушается привычная классификация анемий по цветовому показателю. Анемии подразделяют на группы по различным признакам. Классификацию, как правило, основывают на удобстве и практической значимости для постановки диагноза.
Цветовой показатель покажет степень насыщения эритроцита гемоглобином. У здорового человека ЦП колеблется 0,86 до 1,1. В зависимости от него различают такие анемии:
- Гипохромная анемия, если ЦП меньше 0,86 (по некоторым классификациям ниже 0,8). При таких анализах ставят диагноз талассемия или железодефицитная анемия.
- При нормохромной анемии ЦП колеблется в пределах 0,86 — 1,1. В таком случае у пациента наблюдается постгеморрагическая анемии, гемолитические анемии, апластические анемии, неопластические заболевания костного мозга, внекостномозговые опухоли, анемия из-за снижения темпов выработки и количества эритропоэтина.
- Гиперхромная анемия – при ЦП более 1,1. Такие показатели характерны для фолиеводефицитной анемии, а также витамин B12-дефицитной анемии, для миелодиспластического синдрома.
Проводится в зависимости от выраженности снижения уровня гемоглобина в крови. Классификация по степени тяжести предполагает:
- легкую степень (когда уровень гемоглобина ниже нормы, но остается на уровне более 90 грамм на литр);
- среднюю степень тяжести (когда гемоглобин 70-90 г/л);
- тяжелую степень (когда уровень гемоглобина остается меньше 70 грамм на литр).
Разделение степени анемии по уровню гемоглобина достаточно распространено. Чаще всего именно по гемоглобину подтверждают диагноз, а потом уже проводят более детальную диагностику.
Характеризуется незначительным падением количества гемоглобина в крови. Обычно проявляются следующие симптомы:
- усталость;
- апатия;
- общее недомогание;
- часто развивается у беременных.
Как уже упоминалось выше, анемия 1 степени фиксируется, если уровень микроэлементов в крови ниже нормы, но выше 90 г на л.
Также первая степень заболевания часто поражает маленьких детей, появившихся на свет при многоплодной беременности или недоношенными. У детей постарше – это паразитические заболевания или погрешности в питании. Анемия легкой степени тяжести проще поддается лечению. Обычно рекомендуют соблюдать диету и усиленно питаться. При легкой степени анемии в рацион необходимо включать различные микроэлементы и витамины, в первую очередь – железо и витамин В12.
Анемия 2 степени или средней степени тяжести возникает не только при снижении уровня гемоглобина до 70 г/л. Возникает головокружение и головные боли, одышка, частое сердцебиение, трудности с дыханием. Дети часто болеют, у них наблюдается бледность губ и кожи. При 2 степени анемии может возникнуть кислородное голодание плода, снижается сократительная способность миокарда. Лечение медикаментозное + диета, следует больше находится и гулять по свежему воздуху.
На этой стадии острая анемия несет опасность для жизни человека. При 3 степени наблюдается ломкость ногтей и волос, острое снижение защитных сил организма, онемение конечностей, проблемы с сердцем и сосудами. Ребенок начинает часто болеть.
Особенно тяжелыми будут последствия такого симптома у беременных женщин. Это может негативно отразится на плоде, у матери развиться дистрофические изменения матки и плаценты. Анемия 3 степени лечится в стационаре, с переливанием эритроцитарной массы и другими видами медикаментозного лечения.
Основным признаком такого разделения принято считать количество молодых эритроцитов (ретикулоциты) в периферической картине крови. В норме показатель колеблется от 0,5 до 2%.
- Арегенераторную анемию (например, апластическая анемия) — ретикулоциты отсутствуют.
- Гипорегенераторная (витамин B12-дефицитная анемия или железодефицитная анемия) – ретикулоциты ниже 0,5%.
- Регенераторная или норморегенераторная (постгеморрагическая) — ретикулоциты 0,5 — 2%.
- Гиперрегенераторная (гемолитические анемии) — количество ретикулоцитов больше 2%.
Такая классификация основывается на различных механизмах развития анемии, как патологического процесса.
- Ассоциированные с дефицитом Fe — железодефицитные анемии.
- Гемолитические анемии — связанные с повышенным разрушением эритроцитов.
- Гис-гемолитические анемии — при нарушении кровообразования, протекающего в красном костном мозге.
- Постгеморрагические анемии — при острой или хронической кровопотере.
- В12- и фолиеводефицитные анемии.
Мегалобластные анемии: миокардит гемолитический и пернициозная анемия.
Гипохромия — это общее название для разных форм анемии, при которых цветовой показатель крови, вследствие недостатка гемоглобина, меньше 0,8. Не включена в список нозологических единиц. В анализе крови средний показатель для гемоглобина при таком состоянии меньше 30 пикограмм, а средняя концентрация гемоглобина в эритроците составляет менее 330 г на литр. Меняется не только цвет, но и диаметр (макро- или микроцитоз) и форма. Чаще всего при такого рода патологии красной крови кровяные тельца приобретают вид колец с бледно-красной серединой и темно-красными краями (обесцвечиваются).
Патологические формы эритроцитов могут возникать вследствие:
- Железодефицитной анемии при кровопотерях (геморрагическая), нарушении усвоения железа, при невынашивании плода и т.д.
- Хронического отравления свинцом.
- Талассемии – нарушения нормального процесса синтеза полипептидных цепей в структуре гемоглобина.
- Нарушения синтеза и утилизации порфиринов.
- Гиповитаминоза В6.
- При нарушении обмена вещества, хронических воспалительных процессах неинфекционного и инфекционного генезиса.
Дизэритропоэтическая анемия – целая группа достаточно редко встречающихся заболеваний крови, которые возникают из-за нарушения процессов эритропоэза. При этом, большая часть молодых эритроцитов погибает сразу после образования в костном мозге, а количество нормоцитов значительно снижается. Такого рода расстройство предполагает качественный, количественный, кинетический или совмещенный характер нарушения эритропоэза.
Следует упомянуть такую разновидность заболевания — гиперхромная макроцитарная анемия или малокровие. При котором также наблюдают снижение числа эритроцитов и уровня гемоглобина в единичном объеме крови. Как правило, при такого рода нарушениях параллельно дают сбой сердце и сосуды.
По коду МКБ-10 — D62 острая постгеморрагическая анемия. Такой симптом возникает после кровопотери. Подразделяют на острую постгеморрагическую анемию и хроническую постгеморрагическую анемию. Острая анемия развивается после острых и обильных кровотечений, хронические анемии возникают после длительных несильных кровотечений.
Во время быстрой кровопотери значительно снижается объем циркулирующей крови. В ответ на такое событие происходит компенсаторная реакция, которая заключается в возбуждении симпатического отдела нервной системы и рефлекторного спазма сосудов. Пульс ускоряется и ослабевает. Сосуды кожи и мышц максимально сужаются, кровь приливает к сосудам мозга, коронарным сосудам, что поддерживает снабжение кровью жизненно важных органов. При прогрессировании процесса начинает развиваться постгеморрагический шок.
Характерным клиническим симптомом постгеморрагической анемии является острая сосудистая недостаточность из-за гиповолемии (опустошение сосудистого русла). Внешне это проявляется ортостатическим коллапсом, одышкой, учащенным сердцебиением. В первые минуты кровопотери уровень гемоглобина может даже быть высоким, но после поступления тканевой жидкости в русло сосудов данный показатель снижается, даже если кровотечение уже прекратилось. Цветовой показатель остается в норме, одновременно происходит утрата эритроцитов и железа, нормохромная анемия. Ко вторым суткам увеличивается число ретикулоцитов, максимальных показателей достигает на 4-7 день (гиперрегенераторная анемия).
Диагноз ставят на основании клинических признаков и лабораторных показателей, повышения уровня остаточного азота, если кровотечение происходила в верхних отделах ЖКТ. Самое важное при такой нормохромной анемии – это устранить саму кровопотерю, восполнить устраненный объем крови эритроцитарной массой и гепарином (до 60% утраченной крови), кровезаменителями (5% р-р Альбумина, Раствор Рингера, Реополиглюкин).
При постановке диагноза постгеморрагическая анемия необходимо учитывать сведения о произошедшей острой кровопотере при наличии внешнего кровотечения. После массивного внутреннего диагноз основывают на клинических признаках и, в обязательном порядке, лабораторных пробах (Вебера, Грегерсена). Основанием для постановки диагноза также послужит рост уровня остаточного азота при кровотечении из верхнего отдела пищеварительного тракта.
После того, как кризис и острый период миновали, пациенту назначают препараты железа, витамины группы В, Е и С. Такая терапия производится в течение 6 месяцев. При утрате более 50% от объема циркулирующей крови — прогноз неблагоприятный.
Состояние может развиться в результате снижения поступления в организм фолиевой кислоты или нарушения процессов ее всасывания в пищеварительном тракте. Лечение заключается в дополнительном приеме недостающего вещества.
В отличии от патологии, ассоциированной с недостатком В12, фолиеводефицитная анемия диагностируется значительно реже.
Одной из основных причин фолиеводефицитной анемии является недостаточное содержание фолиевой к-ты в рационе. Следует помнить, что нужно включать в свое ежедневное меню больше зелени и печени. Также на здоровье значительно влияет алкогольная интоксикация, беременность, злокачественные новообразования, некоторые дерматиты, гемолиз.
Такого вида заболевания возникает при нарушении всасывания (например, при целиакии), под действием лекарственных средств, Метотрексата, Триамтерена, противосудорожных средств, барбитуратов, Метформина и др. Значительно повышается нужна организма в фолиевой кислоте после гемодиализа и при болезнях печени.
Также на здоровье оказывает влияние дефицит цианокобаламина и его кофермента – метилкобаламина. В таких условиях не происходит трансформации фолиевой кислоты в коферментную форму. В результате нарушается процесс нормального клеточного деления, начинают страдать клетки кроветворной ткани, которые ранее активно размножались. Тормозятся процессы созревания и размножения эритроцитов, сокращается их продолжительность жизни. Изменения касаются и лейкоцитов, возникает лейкопения и тромбоцитопения.
Далее в результате неправильного митоза появляются гигантские клетки эпителия пищевого канала и развиваются воспалительные процессы в слизистой оболочке, стоматиты, гастриты, эзофагиты, энтерит. Еще больше усугубляется первичное нарушения секреции и процесс всасывания внутреннего фактора, усиливается дефицит витаминов. Возникает порочный круг.
Из-за возникшего недостатка цианокобаламина, в организме начинают накапливаться продукты обмена, которые токсичны для нервных клеток. В волокнах тем временам начинают синтезироваться жирные кислоты уже с искаженной структурой. Наблюдается плавное изменение качества клеток спинного мозга и поражаются периферические и черепные нервные сплетения, развивается неврологическая симптоматика.
При таком виде анемии, как правило симптомы у пациента наблюдаются стандартные: высокая утомляемость, учащенное сердцебиение, бледность ногтей и губ, ярко-красный язык. На начальных стадиях отследить признаки поражения НС и ЖКТ практически невозможно. По результатам обследования выявляется гиперхромная макроцитарная анемия, лейкопения, тромбоцитопения. А лечение с помощью витамина В12 не вызывает улучшений лабораторных показателей. Диагноз можно подтвердить с помощью определения уровня сывороточной фолиевой к-ты и в эритроцитах. В норме ее содержание составляет от 100 до 450 нг/л. При фолиеводефицитной анемии концентрация фолиевой кислоты в эритроцитах значительно снижается.
Если проводят анализ периферической картины крови, то отмечают гиперхромную (макроцитарную) анемию при общем снижении уровня гемоглобина и эритроцитов. Непрямой билирубин меняется редко.
В качестве профилактики и для лечения фолиеводефицитной анемии должны назначить фолиевую кислоту в дозе 1 мг/сут, внутрь. Если есть необходимость врач может увеличить дозу. Улучшения должны наступать через в течение 3-4 суток, в том числе проходить должны и неврологические симптомы. В противном случае приходится говорить о недостатке именно витамина В12, а не фолиевой к-ты.
Обязательно проводить профилактику недостатка фолиевой к-ты у беременных женщин и пациентов, принимающих некоторые группы лек. препаратов. Для профилактики назначают 5 мг вещества в сутки.
Относится к виду витаминодефицитных (мегалобластных) анемий, которые возникают при недостаточном поступлении в организм витамина В12 (цианокобаламина). В результате В12-дефицитной анемии поражается кроветворная функция, нервная и пищеварительная система. В отличие от фолиеводефицитной В12-анемии, она развивается в преклонном или старческом возрасте, чаще всего у мужской половины. Проявляется заболевание постепенно.
Человек, страдающий В12-дефицитной анемией, будет испытывать общие симптомы: слабость, пониженная работоспособность, одышка, головокружение, жжение за грудиной, боли в ногах и языке, парестезии, шаткость походки. Такие симптомы будут сочетаться с желтушным цветом кожи, глосситом, незначительным увеличением печени и селезенки, негромким систолическим шумом, глухостью тонов сердца. Нередко поражается и нервная система, развивается комбинированный склероз или фуникулярный миелоз, нарушается чувствительность, полиневрит, атрофия мышц, паралич нижних конечностей.
Развивается анемия из-за недостатка витамина В12, из-за несбалансированности питания, голодания, алкоголизма и отсутствия аппетита. Также синдром может наблюдаться на фоне мальабсорбции, целиакии, изменений в слизистой оболочке кишечника, спру, лимфомы кишечника, болезни Крона, регионарного илеита, из-за длительного приема противосудорожных препаратов.
Вероятность развития В12-дефицитной анемии будет выше при беременности, гемолитической анемии, псориазе, эксфолиативном дерматите. При приеме алкоголя, препаратов–антагонистов фолатов, врождённых нарушениях метаболизма и атрофическом гастрите также может наблюдаться это заболевание.
Как правило, поставить диагноз В12-дефицитная анемия можно без особых проблем. Это может сделать гематолог, невролог, нефролог или гастроэнтеролог по результатам общего и биохимического анализа крови, наличию метилмалоновой кислоты. Также проводят УЗИ брюшной полости и исследование всасывания витамина В12 с помощью радиоизотопов. По показаниям можно провести аспирационную биопсию костного мозга.
Лечение проводят витамином В12 внутримышечно. В течение 1-1,5 месяцев используют поддерживающую дозу. Если уровень гемоглобина упал менее 60 г на л, при нарушении гемодинамики и угрозе анемической комы проводят трансфузии эритроцитов.
В качестве профилактики при заболеваниях, сопровождающихся нарушением всасывания витамина В12, а также после операций нужно под контролем содержания витамина В12 в моче и крови применять профилактические и лечебные курсы витаминотерапии.
Что это такое простыми словами? Это процесс ускоренного разрушения красных кровяных телец, он сопровождается значительным ростом уровня прямого билирубина в крови. Заболевание встречается достаточно редко.
Общим признаком можно считать ускоренное разрушение эритроцитов, сопровождающееся с одной стороны анемией и повышенным образованием продуктов распада эритроцитов, а с другой – реактивно усиленным эритропоэзом.
Аутоиммунная гемолитическая анемия, как правило вызвана генетическими дефектами мембран эритроцитов, что вызывает их повышенное разрушение. Аутоиммунное заболевание и гемолиз эритроцитов при приобретенных анемиях наступает под действием внутренних факторов или каких-либо факторов окружающей среды.
На развитие иммунных гемолитических анемий влияют посттрансфузионные реакции, вакцинации, прием некоторых препаратов (сульфаниламидов, анальгетиков, противомалярийных лекарств, производных нитрофуранового ряда). Также такой симптом может проявляться при гемобластозах, аутоиммунных патологиях (НЯК, СКВ), инфекционных болезнях (мононуклеоз, вирусная пневмония, сифилис, токсоплазмоз).
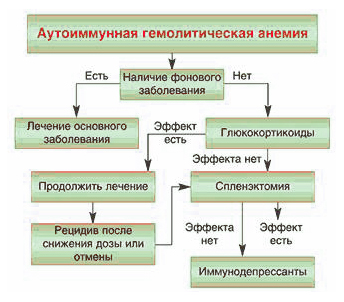
Гемолитические анемии можно поделить на два обширные группы заболеваний: приобретенные и врожденные.
Формы наследственных заболеваний:
- Эритроцитарные мембранопатии, такие как анемия Минковского-Шоффара или болезнь Минковского-Шоффара (микросфероцитоз), акантоцитоз, овалоцитоз, вызванные аномалиями в структуре мембран эритроцитов. Отметим, что сфероцитоз – самый распространенный вид среди патологий (микросфероцитоз).
- Энзимопении, вызванные дефицитом некоторых ферментов (пируваткиназа, глюкозо-6-фосфатдегидрогеназа).
- Гемоглобинопатии, возникшие из-за качественных нарушений структуры гемоглобина или изменения соотношения его нормальных форм.
Приобретенные гемолитические анемии делят на:
- Приобретенные мембранопатии (шпороклеточная анемия, болезнь Маркиафавы-Микели).
- Изо- и аутоиммунные, вызванные действием антител.
- Токсические, возникшие вследствие воздействия ядов, токсинов или других хим. агентов.
- Анемии, ассоциированные с механическими повреждениями в структуре эритроцитов.
Рост концентрации остаточных следов после распада эритроцитов в организме внешне будет проявляться желтухой лимонного оттенка. Также будет наблюдаться повышение концентрации в крови непрямого билирубина и железа. Отмечается уробилинурия и плейохромия кала и желчи. При внутрисосудистом гемолизе дополнительно развивается гипергемоглобинемия, гемоглобинурия, гемосидеринурия. Об усилении эритропоэза говорит ретикулоцитоз и полихроматофилия в составе периферической крови, либо эритронормобластоз костного мозга.
Если у пациента не микросфероцитарная анемия, обусловленная наследственным сфероцитозом или эллиптоцитозом, то необходимо, прежде всего проводить адекватную терапию заболевания, вызвавшего причину гемолитической анемии.
Для лечения обычно применяют:
- медикаменты (например, Десферал);
- спленэктомию;
- трансфузию эритроцитов при кризе;
- ГСК при аутоимунных заболеваниях в средней дозировке.
Апластическая анемия — заболевание, которое относят к категории миелодисплазий. При таком заболевании происходит резкое угнетение или прекращение роста и созревания клеток в костном мозге, еще называемое панмиелофтизом.
Типичными симптомами болезни являются: лейкопения, анемия, лимфопения и тромбоцитопения. Сам термин впервые появился в начале 20 века. Такое заболевание имеет достаточно тяжелое течение и без лечения (в том числе медикаментозного препаратом Атгам) имеет неблагоприятный прогноз.
Очень долго эту болезнь рассматривали в качестве синдрома, объединяющего различные патологические состояния в костном мозгу. В настоящее время термин «апластическая анемия» выделяется в качестве самостоятельной нозологической единицы. Его следует четко разграничивать от синдрома гипоплазии в кроветворной системе.
Апластическая анемия может быть вызвана различными причинами:
- химическими агентами, бензолом, солями тяжелых металлов и так далее;
- ионизирующим излучением;
- приемом некоторых лекарств, цитостатиков, НПВС, Анальгина, Мерказолила, Левомицетина;
- вирусами;
- наличием других аутоиммунных заболеваний.
Также существует форма апластической анемии, передающейся по наследству — анемия Фанкони. Лечение болезни заключается в приеме иммунодепрессантов и проведении пересадки костного мозга.
При серповидно-клеточной анемии человека происходит нарушение строения белка гемоглобина, он приобретает нетипичное кристаллическое строение, в виде серпа. Такую форму называют S-гемоглобином. Заболевание связывают с мутацией НВВ-гена, из-за которой в костном мозге начинает синтезироваться аномальный вид S-гемоглобина, в шестом положении в В-цепи вместо глутаминовой кислоты находится валин. Происходит полимеризация S-гемоглобина, образуются длинные тяжи, эритроциты приобретают форму серпа.
Тип наследования серповидноклеточной анемии – аутосомно-рецессивный при неполном доминировании. У гетерозиготных носителей в эритроцитах находится примерно равное количество гемоглобина А и S. Носители сами не болеют, а выявить серповидные эритроциты можно случайно при проведении лабораторного обследования. Симптомы же могут и вовсе не проявляться. Иногда такие люди начинают ощущать недомогание при гипоксии или тяжелом обезвоживании.
У гомозигот в крови есть только гемоглобин S, заболевание протекает достаточно тяжело. У таких больных высокий уровень степени разрушенных эритроцитов в селезенке, значительно короче срок жизни, часто проявляются признаки хронической недостаточности кислорода.
Такой вид анемии достаточно распространен в регионах, где высокий уровень заболеваемости малярией. У таких больных выше устойчивость к разным штаммам малярийного плазмодия. Поэтому столь вредные аллели часто проявляются у жителей Африки.
Симптомы сильно различаются и их можно наблюдать у детей уже с 3-х месячного возраста. Анемия может приводить к потере сознания, меньшей выносливости, вызвать желтуху. У младенцев наблюдается худоба, слабость, искривление конечностей, удлиненность туловища, изменения в строение черепа и зубов. Также у больных детей повышенная склонность к развитию сепсиса. У подростков наблюдают задержку развития на 2-3 года. Женщины, как правило, способны к зачатию и рождению ребенка.
Мегалобластная анемия (болезнь Аддисона-Бирмера, В12-дефицитная, пернициозная) — заболевание, вызванное недостатком фолиевой кислоты или витамина В12. Происходит это из-за недостатка веществ в пище или при заболеваниях пищеварительного тракта. Также мегалобластическая анемия может возникнуть при врожденных нарушениях процессов синтеза ДНК, приобретенных патологиях и из-за приема некоторых лекарств (антиметаболитов, противосудорожных).
При постоянном недостатке фолиевой кислоты и В12 развивается хроническая анемия, эритроциты изменяют свою форму и размеры. Более легкие стадии иногда протекают бессимптомно, далее начинают уже проявляться внешние признаки. Такое дефицитное состояние часто также называют пернициозной анемией. Заболевание получило статус анемии хронических заболеваний, так как проявляется у пациентов после 60 лет и у перенесших гепатит больных, при энтерите и раке кишечника. Подробнее о такого рода анемиях описано выше.
Развивается вследствие эндогенного В12-авитаминоза, вызванного атрофией желез фундального отдела желудка, которые в норме должны вырабатывать гастромукопротеин. В результате нарушаются процессы всасывания витамина В12 и возникает злокачественная анемия «пернициозного» типа. Чаще всего такой диагноз ставят в возрасте от 50 лет.
Болезнь сопровождается нарушениями нервной, сердечно-сосудистой, кроветворной и пищеварительной системы. Больные обычно жалуются на одышку, общую слабость, отечность ног, боли в сердце, «мурашки» по стопам и кистям, жгучие боли в языке и шаткость походки. По лабораторным показателям наблюдают анемию гиперхромного типа, лейкопению, тромбоцитопению.
Сидеробластная анемия также имеет название сидероахрестическая анемия (САА), железорефрактерная, железонасыщенная или сидеробластическая. Это патологическое состояние нарушений процесса синтеза микроэлементов и кроветворения, чаще всего – железа. Эритроциты содержат малое количество железа, из-за того, что микроэлемент активно расходуется костным мозгом и начинает накапливаться во внутренних органах. Заболевание развивается на фоне недостаточного содержания протопорфирина.
Выделяют две формы болезни:
- пиридоксин-зависимую, возникающую вследствие дефицита пиридоксальфосфата;
- пиридоксин-резистентную, развивающуюся из-за ферментного дефекта (дефицита гемсинтетазы).
Приобретенные формы сидероахрестической анемии чаще наблюдаются в пожилом возрасте, но болезнь не наследуется от родителей. Часто такого рода анемия развивается, как побочный эффект от лечения препаратами от туберкулеза или истощении пиридоксальфосфата при отравлении свинцом, при алкоголизме, миелопролиферативных болезнях крови, кожной порфирии. Встречаются и идиопатические формы САА.
Это редкое, передающееся по наследству заболевание. Анемия Фанкони встречается у 1 из 350000 детей. Наибольшее распространение заболевание получила среди евреев-ашкеназов и жителей Южной Африки.
Возникает из-за наличия дефектов в белковых кластерах, отвечающих за процесс репарации ДНК. Для заболевания характерная высокая ломкость у хромосом, наличие у пациентов старше 40 лет миелоидного лейкоза и апластической анемии.
Для новорожденных с таким недугом характерны врожденные дефекты развития, необычная пигментация, низкорослость, аномалии развития скелета и некоторые неврологические симптомы (косоглазие или недоразвитость одного из глаз, глухота, умственная отсталость), аномалии развития внутренних органов. К сожалению, в среднем такие пациенты живут не более 30 лет.
У диагноза неуточненная анемия код по МКБ-10 D64.9. Это первичный диагноз, который уточняется после проведения обследования у врача, так как он является вторичным признаком какого-либо основного заболеваний. Прежде всего следует исключить возможность кровопотери вследствие травм, хирургических вмешательств, внутренних кровотечений, далее проводится лабораторная диагностика.
По международной классификации МКБ-10 для миелодиспластического синдрома:
- D46.0 Рефрактерная анемия без сидеробластов, так обозначенная;
- D46.1 Рефрактерная анемия с сидеробластами;
- D46.2 Рефрактерная анемия с избытком бластов;
- D46.3 Рефрактерная анемия с избытком бластов с трансформацией;
- D46.4 Рефрактерная анемия неуточненная;
- D46.7 Другие миелодиспластические синдромы;
- D46.9 Миелодиспластический синдром неуточненный.
Приставка «рефрактерная» означает устойчивость заболевания к приему витаминов, препаратов железа, соблюдению диеты. Чаще всего такого вида анемия является наиболее распространенным видом миелодиспластического синдрома. Из-за нарушений процессов созревания бластов в крови значительно понижено содержание гемоглобина, проявляются признаки острого лейкоза. Примерно у 40% пациентов с миелодиспластическим синдромом проявляется рефрактерная анемия. Чаще всего гемоглобинопатия такого рода развивается у пациентов от 50 лет.
Заболевание можно считать промежуточным этапом между рефрактерной анемией и острым лейкозом. Как правило, болезнь проявляется снижением уровня гемоглобина и общей слабостью. Если у больного не нашли других причин для таких изменений картины крови, то основной задачей врачей становится дообследовать пациента и максимально затормозить наступление острого лейкоза.
Что за болезнь талассемия? Это наследуемое по рецессивному типу заболевание, развивающееся из-за снижения синтеза полипептидных цепей в структуре гемоглобина. В зависимости от того, какой мономер перестал нормально синтезироваться различают альфа-, бета-талассемию и дельта-талассемию. Также болезнь классифицируют по степени клинических проявлений, разделяют на тяжелую, легкую и среднюю.
Альфа-талассемия ассоциирована мутациями в генах HBA2 и HBA1. Альфа-цепь кодируется четырьмя локусами и, в зависимости от количества аномальных – различают разные степени тяжести заболевания. Симптомы и течение гемоглобинопатии варьируют от легкой до тяжелой степени гипохромной микроцитарной анемии.
Бета-талассемия существует в двух наиболее распространенных вариантах: малая (minor) и CD8(-AA) – большая (наиболее тяжелая форма заболевания). Анемия развивается из-за мутаций обоих аллелей бета-глобина, когда гемоглобин А начинает вытесняться гемоглобином F. Обычно малая талассемия имеет легкое течение и лечение не требуется.
Согласно Википедии, анемия Даймонда-Блекфена представляет собой наследственную форму красноклеточной аплазии с точно не выявленным типом наследования. Предполагают, что заболевание имеет аутосомно-доминантный тип наследования, который встречается у одной четвертой всех больных. У таких больных обычно проявляются проявления анемии в течение первого года жизни, слабость, бледность, повышенная утомляемость, снижение количества эритроцитов в плазме крови.
Диагноз ставят по общему анализу крови, показателям уровня эритропоэтинов, микроскопии и биопсии костного мозга. Лечению болезнь поддается плохо, назначают ГКС, гемотрансфузии.
Повышение концентрации свободного гемоглобина в плазме крови называют гемоглобинемией. Заболевание возникает вследствие приобретенных и врожденных заболеваний эритроцитов, при повышенном их разрушении. Симптом может проявиться после неправильно проведенного переливания крови, из-за воздействия паразитарных и инфекционных факторов (сепсиса, вирусной инфекции, малярии), после сильного переохлаждения, отравления ядами, бензином, уксусной кислотой, грибами. Проявляется внешне это гиперемией кожных покровов и слизистой, повышением концентрации гемоглобина и билирубина в крови.
Существуют настолько разные причины заболевания, что выявить некоторые из них можно только после проведения лабораторных исследований. Часто бывает, что показатели гемоглобина в норме, а железо низкое. Недуг может наблюдаться при недостатке витаминов группы В, фолиевой к-ты. Малокровие у человека возникает вследствие различных травм и обильных внутренних или внешних кровотечений.
Такой диагнозе женщине могут поставить, если уровень гемоглобина в крови падает ниже 120 г на литр. В целом у женщин, из-за некоторых физиологических особенностей выше склонность к данному недугу. Ежемесячные кровопотери во время менструации, когда в течение недели женщина может потерять до 100 мл крови, беременность, лактация и снижение концентрации ферритина – самые распространенные причины анемии. Часто заболевание развивается из-за психосоматики, когда у женщины депрессивное состояние, она плохо питается, редко выходят на свежий воздух, не занимается спортом или во время климакса, когда происходят глобальные изменения в физиологии женщины.
У мужской половины данный диагноз ставится, если уровень гемоглобина снизился менее 130 г на л. По статистике заболевание возникает реже. Чаще всего из-за каких-то хронических заболеваний, нарушений работы органов и их систем (язвенная болезнь, геморрой, эрозии в кишечнике, доброкачественные и злокачественные новообразования, паразиты, апластическая анемия). Существуют и генетические причины (анемия Фанкони) заболевания, которые не связаны с полом.
Малокровие проявляется по-разному, в зависимости от конкретного вида заболевания. Известно, что при малокровии у человека уменьшается количество эритроцитов, изменяется их строение, содержание железа в крови, падает гемоглобин. Ткани человека испытывают недостаток кислорода и это оказывает влияние на общее состояние и внешний вид человека.
Общие признаки анемии, следующие:
- значительно сниженная работоспособность, общая слабость;
- раздражительность, повышенная утомляемость, сильная сонливость;
- шум в ушах и головные боли, «мушки» перед глазами, головокружения;
- дизурия;
- непреодолимое желание полакомится мелом или известью;
- постоянная одышка;
- тонкие и ломкие волосы, ногти, сухая, неэластичная кожа;
- стенокардия, низкое артериальное давление;
- шум в ушах и частые обмороки;
- изменение цвета стула, желтуха, бледность;
- ломота в теле и суставах, мышечная слабость.
Также существуют специфические симптомы малокровия, характерные для той или ной разновидности:
- Железодефицитная анемия. Для данного диагноза характерная парорексия, у пациента появляется сильное желание жевать мел, землю, бумаги и прочие несъедобные материалы. Можно также выделить койлонихию, трещинки в уголках рта, заеды, воспаленный язык. Иногда может повыситься температура до субфебрильной.
- Основным симптомом В12-дефицитной анемии можно назвать покалывание в конечностях, нестойкость походки, скованность и стесненность в движениях, низкое чувство осязания. У пациента снижаются когнитивные способности, могут возникать галлюцинации. В крайне тяжелых случаях может развиться паранойя или шизофрения.
- Характерным симптомом серповидноклеточной анемии может быть слабость, приступообразные боли в брюшной полости и суставах.
- При отравлении свинцовыми отходами у пострадавшего наблюдаются характерные темно-синие линии на деснах, тошнота и болезненные ощущения в области живота.
- Хроническое разрушение эритроцитов может быть симптомом злокачественной опухоли. В таком состоянии развивается проступающая желтуха, язвы и ссадины на ногах, покраснение мочи. Часто образуются камни в желчном пузыре.
Поставить точный диагноз гемолитическая, мегалобластная или апластическая анемия можно после проведения дифференциальной диагностики. Диф. диагностика проводится по соответствующим таблицам и результатам лабораторных исследований.
источник