Заболевание почек – серьезная патология во всем мире. Так как почки, прежде всего выделительный орган, благодаря которому из организма выводится мочевина и вода. Разберем понятие амилоидоз более подробно.
Амилоидоз почек – это отложение в почечной ткани амилоида. Амилоид — сложный мукополисахарид. Какие симптомы, свойственны данному заболеванию?
Симптомы нарастают постепенно. Сначала более незначительные признаки в виде изменения цвета кожи, плохой анализ мочи. Затем возникают отеки. Асцит и гидроторакс – затронута наиболее обширная часть отечности.
Для более подробной информации читайте сайт: bolit.info
В развитии амилоидоза почек выделяют несколько стадий. На первой стадии течения заболевания признаки не выявлены. На второй стадии заболевание протекает вторично, то есть на фоне других болезней. Какие заболевания могут вызвать амилоидоз почек?
Прежде всего, болезнь протекает на фоне иммунодефицита. Это состояние наиболее располагает к заболеванию. На втором месте онкологические заболевания, которые под влиянием химиотерапии, истощают организм. На третьем месте инфекционные болезни, допустим, туберкулез.
Часто амилоидоз почек развивается на основе другого заболевания. Поэтому для предупреждения данной болезни нужно устранить основную причину. Если причина в туберкулезе, лечите палочку Коха. При малярии устраняйте эту тяжелую инфекционную болезнь.
Если, проходите, курс гемодиализа, сократите, по возможности, длительность данного процесса. Бывает, причина в старости, при которой пожилых людей поражает болезнь слабоумия. В таком случае, болезнь может прогрессировать, следствием будет – амилоидоз почек. Профилактика, и в том, и в ином случае, необходима для любого человека.
Как и при любом другом заболевании, назначают лабораторные методы исследования. А именно – кровь, моча. В моче прослеживаются признаки заболевания почек. В крови – воспалительный процесс, лейкоцитоз, анемия.
Однако, лабораторные методы недостаточны в диагностике данного заболевания. Эти исследования только на начальной стадии болезни. Если же заболевание имеет затяжной характер, то проводят дополнительные исследования.
Исследование кала для определения болезни по консистенции испражнения. Если кал жидкий, то это амилоидоз почек. Проводят ультразвуковое исследование, при котором наблюдаются изменения в печени и селезенке. Рентген желудочно-кишечного тракта. При этом наблюдаются изменения в пищеводе. Также, необходимо отметить, что при исследованиях видно увеличение почек.
Как было сказано выше, болезнь может быть в пожилом возрасте. Среди людей среднего возраста она тоже встречается. Как вылечить болезнь на вторичной стадии? Важен рацион взрослого человека. Данная диета касается взрослых людей.
— значительное количество витаминов
Используют медикаментозную терапию. Назначают гемодиализ. Мочегонные препараты для уменьшения отеков. Препараты, снижающие давление. Переливание плазмы или же пересадка почки. Стоит учесть, что лечение отеков, переливание плазмы недостаточны, это дает лишь временный эффект. Если же амилоидоз почек вызван инфекционной болезнью, то важно устранить ее распространение и дальнейшие рецидивы.
Дети болеют реже. Для них характерен вторичный амилоидоз почек. Но, встречаются случаи, когда ребенок заболевает по вине своей матери. Если женщина вела нездоровый образ жизни, беременность протекала тяжело. Хронические заболевания у детей также провоцируют данную болезнь. Какова клиническая картина?
Клиника напоминает симптомы заболевания почек, помимо этого беспокойное, апатичное состояние ребенка, отеки. Диагностика болезни у детей затруднена.
Лечение обычно проводят противоопухолевыми препаратами, а именно – цитостатические лекарственные вещества. Препараты природного происхождения еще не используются в современной практике. Печально, но уровень сегодняшней медицины развивается быстро. Кто знает, что готовит нам и нашим детям век модернизированной медицины.
Амилоидоз почек можно прогнозировать. Прогноз будет неблагоприятным при условии игнорирования основного заболевания. Также встречаются осложнения. При осложнениях назвать прогноз благоприятным практически невозможно. С чем это связано?
Если состояние больного осложнено различными кровоизлияниями. Вследствие снижения иммунитета – присоединение инфекции. Много зависит от своевременности. Чем быстрее вы обратитесь к врачу, тем больше возможности вылечиться и предотвратить неблагоприятные последствия.
Если обстоятельства складываются в пользу больного, то исход заболевания будет хороший. Для этого пройдите курс лечебной терапии. При трансплантации почки продолжайте проводить реабилитационные мероприятия.
Обратитесь к нефрологу! Данный специалист наиболее точно поставит диагноз, назначит курс медикаментозной терапии. Конечно же, определит рацион при амилоидозе почек. Если прогноз заболевания хороший, то, не сомневайтесь, исходом будет – выздоровление.
Жизнь человека во многом зависит от состояния организма. Здоровье – фактор продолжительности жизни. Но, если заболевание вызвало необратимые последствия, то продолжительность жизни становится короче. По статистике, небольшой процент людей, при осложнениях касающихся сердечной деятельности, доживает до пожилого возраста.
Факт, что при амилоидозе почек возможны летальные исходы. Это вызвано, прежде всего, патологией сердечнососудистой системы. Отеки как симптом данного заболевания, вызывают нарушение сердечного ритма. Следствием является более тяжелые сердечные болезни. Так что обращайтесь к врачу раньше времени развития вторичной стадии заболевания! Будьте здоровы!
источник
Основные направления лечения амилоидоза:
- уменьшение синтеза белков-предшественников;
- рассасывание отложений амилоида;
- симптоматическое лечение проявлений амилоидоза.
Теоретическим обоснованием современного лечения амилоидоза является так называемая концепция «предшественник — продукт», в основе которой лежит идея о том, что дальнейший рост амилоидных депозитов останавливается по мере прекращения поступления необходимого белка-предшественника. Соответственно, при АА-амилоидозе необходимо уменьшение циркулирующего SAA, для AL — легких цепей иммуноглобулинов, для ATTR — мутантного транстиретина, для диализного амилоидоза — бета2-микроглобулина.
Основой стратегии лечения АА-амилоидоза является активное воздействие на основное заболевание с целью снижения амилоидогенного стимула — уменьшения уровня циркулирующего белка-предшественника SAA. Интенсивное лечение, позволяющее снизить уровень SAA менее чем до 4-10 мг/л, может остановить амилоидогенез и индуцировать постепенное улучшение почечной функции и исчезновение амилоидных депозитов. При хронических инфекционных заболеваниях и туберкулезе это успешная антибактериальная терапия, при остеомиелите — хирургическое лечение, при воспалительных ревматических заболеваниях — эффективное и стойкое подавление воспалительного процесса.
Применение колхицина позволило значительно улучшить прогноз у пациентов со средиземноморской лихорадкой. Этот препарат не только урежает рецидивы болезни, но и существенно снижает риск развития амилоидоза. Механизм действия препарата до сих пор точно не установлен. Элиминация амилоидных депозитов вероятнее опосредована супрессией воспалительного ответа и выработки SAA, чем воздействием на другие звенья амилоидогенеза. Колхицин применяется также для лечения амилоидоза у больных с РА, АС, воспалительными заболеваниями кишечника (ВЗК), ПсА. Имеются данные как о положительном эффекте в виде уменьшения признаков нефротического синдрома и азотемии, так и о безуспешном применении этого препарата.
У больных с воспалительными ревматическими заболеваниями, осложненными АА-амилоидозом, определенный эффект дают алкилирующие цитостатики, в частности лейкеран. Препарат оказывает иммунодепрессивное, антипролиферативное, антивоспалительное воздействие. Лейкеран с успехом применялся у больных с АА-амилоидозом при РА, ПсА, в том числе с псориатическим спондилоартритом. Лечение лейкераном в дозе 2-8 мг/сут приводило к стабилизации амилоидных депозитов, уменьшению проявлений нефротического синдрома и увеличивало 5-летнюю выживаемость пациентов до 80-90%.
Диметилсульфоксид (ДМСО) при пероральном приеме продемонстрировал положительный эффект у больных с амилоидозом при РА, АС, ПсА на разных стадиях заболевания, препятствуя его развитию и способствуя регрессии уже имеющихся депозитов. Примерно через 3 мес приема у больных отмечалось уменьшение протеинурии, повышение клубочковой фильтрации, снижение азотемии. Этот метод лечения отличается хорошей переносимостью, редко возникают тошнота и отвращение к запаху препарата. В этом случае рекомендуется разводить препарат фруктовым соком или мятой.
Определенные надежды в лечении АА-амилоидоза связаны с развитием и внедрением в практику средств биологической терапии, в частности ингибиторов ФНО-а. Теоретическим обоснованием этого лечения являются данные о том, что ФНО-а индуцирует синтез SAA в гепатоцитах в процессе острофазового ответа, способствуя формированию амилоидных фибрилл. Первым препаратом такого типа, нашедшим применение при ряде ревматических заболеваний и амилоидозе, стал инфликсимаб (ремикейд). Инфликсимаб с высокой специфичностью связывает как циркулирующий, так и фиксированный на клеточных мембранах ФНО-а. Имеются обнадеживающие данные о позитивном воздействии инфликсимаба на течение АА-амилоидоза у пациентов с различными воспалительными ревматическими заболеваниями: РА, АС, ювенильным хроническим артритом (ЮХА), артритом при ВЗК. У большинства пациентов при лечении амилоидоза отмечалось быстрое и стойкое снижение протеинурии, увеличение клубочковой фильтрации, а также прекращение формирования амилоидных депозитов по данным сцинтиграфии с меченным 123 I SAP. Инфликсимаб дает эффект также у больных с ССЛ, воздействуя на частоту и выраженность приступов и предотвращая развитие амилоидоза.
Перспективы лечения и профилактики АА-амилоидоза, ассоциированного с другими аутовоспалительными синдромами (TRAPS, CAPS), также связаны с генно-инженерными биологическими препаратами. Существенно улучшить прогноз у пациентов с CAPS, причиной гибели которых является АА-амилоидоз, позволило применение рекомбинантного растворимого рецептора к IL-1 — анакинры. Имеются данные об эффективности этанерцепта при синдроме TRAPS. Этанерцепт продемонстрировал эффективность при лечении АА-амилоидоза у больных ревматоидным артритом, анкилозирующим сподилитом. Ha фоне лечения отмечалось не только улучшение клинических параметров воспалительного процесса, но и снижение протеинурии и сывороточного креатинина.
Опубликовано несколько сообщений об успешном лечении АА-амилоидоза при ревматических заболеваниях тоцилизумабом (актемрой). Препарат представляет собой рекомбинантное гуманизированное моноклональное антитело к человеческому рецептору IL-6. При назначении в дозе 8 мг/кг каждые 4 недели у пациентов с РА, ЮХА и амилоидозом кишечника наблюдалось быстрое, иногда «драматическое» улучшение в виде прекращения диареи, уменьшения и даже исчезновения амилоидных депозитов, снижения активности основного заболевания.
| Препарат | Доза | Механизм действия | Показания |
| Колхицин | 2-4 мг/сут | Антимитотическое, антипролиферативное действие, тормозит образование амилоидных фибрилл | ССЛ, РА, АС, ПсА, ВЗК |
| Лейкеран | 2-8 мг/сут | Противовоспалительное, |
антипролиферативное
Среди перспективных подходов к лечению амилоидоза-АА следует отметить непосредственное воздействие на фибриллообразование и формирование амилоидных депозитов. Таким свойством обладает ряд низкомолекулярных сульфатов, конкурирующих с гликозаминогликанами и препятствующих их взаимодействию с амилоидным белком. Таким образом предотвращается образование амилоидных фибрилл. В 2007 г. опубликованы данные многоцентрового исследования оценки эффективности и безопасности препарата эпродизат (фибриллекс) у пациентов с АА-амилоидозом и почечной недостаточностью. Это отрицательно заряженная сульфатированная молекула, напоминающая по структуре гепарин-сульфат. Конкурируя с гепарин-сульфатом, фибриллекс предотвращает сшивание амилоидных полипептидных цепей, их полимеризацию и депонирование. Продемонстрировано снижение прогрессирования ХПН на фоне лечения фибриллексом. Разрабатываются также препараты, способствующие димеризации молекулы SAP и быстрому клиренсу комплексированного протеина.
До 1990 г. эффективного лечения амилоидоза-AL не было. Первой схемой, позволившей увеличить продолжительность жизни больных с 6-9 до 18 мес, было предложенное в 1996 г. сочетание мелфалана и преднизолона. Однако данное лечение оказалось эффективным лишь у 25% пациентов. Кроме того, продолжительность эффекта не превышала 6-8 мес. В настоящее время предпочтительной терапией считается применение высоких доз мелфалана (200 мг на 1 м 2 поверхности тела) в сочетании с трансплантацией аутологичных стволовых клеток крови. Данная схема лечения дает высокий гематологический и органный ответ — 83%, выживаемость больных составляет 7,8 лет, 5-летняя выживаемость — 61%. Одна из основных проблем этой схемы — высокий показатель ассоциированной с терапией смертности — 12-13%. Существуют и другие схемы лечения AL-амилоидоза: средние дозы мелфалана в сочетании с трансплантацией стволовых клеток, высокие дозы мелфалана в сочетании с дексаметазоном, талидомид с дексаметазоном. В небольших исследованиях продемонстрирована эффективность 2-3-х циклов индукционной терапии перед основной схемой с использованием винкристина, доксорубицина и дексаметазона. В таблице представлены основные схемы лечения AL-амилоидоза.
Основные схемы лечения AL-амилоидоза
|
| Примечание. HDM (high-dose melphalan) — высокие дозы мелфалана; IDM (intermediate dose melphalan) — средние дозы мелфалана; ASCT (autologous stem cell transplantation) — трансплантация аутологичных стволовых клеток; MDex (melphalan plus dexamethasone) — мелфалан + дексаметазон; HD-Dex (high-dose dexamethasone) — высокие доз мелфалана + дексаметазон; МР (melphalan plus prednisone) — мелфалан + преднизолон. |
При миелома-ассоциированном амилоидозе в последние годы применяются схемы, включающие бортезомиб, — ингибитор протеасом, обладающий избирательной цитотоксичностью в отношении опухолевых клеток. Бортезомиб назначается как в качестве монотерапии, так и в комбинации с дексаметазоном, доксорубицином в разных режимах в качестве индукционной терапии.
Важной составляющей лечения амилоидоза является симптоматическая терапия. Она позволяет улучшить качество жизни пациентов и создать своеобразный мост до наступления эффекта патогенетической терапии, хотя в ряде случаев, особенно при полисистемном поражении, достигнуть успеха достаточно сложно.
| Клинический синдром | Лечение амилоидоза |
| Амилоидная кардиопатия | Диуретики, антиаритмики (амиодарон), дезагреганты, имплантация ЭКС. Трансплантация сердца |
| Амилоидная нефропатия | Гемодиализ, трансплантация почки |
| Амилоидоз кишечника | Антидиарейные препараты, антибактериальные препараты, пробиотики |
| Амилоидная нейропатия | Витамины группы В, вазоактивные препараты, антиоксидантная терапия, при нейропатической боли — нейротропные средства (габапентин) |
| Амилоидная артропатия | НПВП |
| Синдром запястного канала | Хирургическая декомпрессия срединного нерва |
| Легочные узловые депозиты | Хирургическое лечение амилоидоза |
Основным подходом к лечению ATTR-амилоидоза в настоящее время является трансплантация печени — органа, в котором синтезируется транстиретин. Трансплантация печени приводит к улучшению вегетативных функций (сфинктерных, кишечных расстройств, ортостатической гипотонии). Однако при развитии периферической нейропатии с необратимыми дегенеративными нарушениями нервных стволов добиться регресса не удается.
В последние годы с успехом разрабатываются медикаментозные методы лечения амилоидоза. В основе одного из них лежат стабилизация нативной структуры белка-предшественника и предотвращение его трансформации в белок амилоида. Таким свойством обладает находящийся на стадии исследования препарат дифлюнизал, молекула которого соединяется с тироксинсвязывающим концом TTR-тетрамера, стабилизируя протеин и препятствуя его тенденции к трансформации в складчатую амилоидную конфигурацию.
На животных моделях продемонстрирована способность доксициклина провоцировать деградацию амилоидных фибрилл при ATTR-амилоидозе.
Определенные перспективы в лечении AH-амилоидоза связываются с созданием новых диализных мембран, пропускающих AH-микроглобулин, а также с использованием прямой гемоперфузии и гемодиафильтрации. Своевременная трансплантация почки позволит избежать аккумуляции AH-микроглобулина и уменьшить риск развития амилоидоза. Однако у пациентов с уже развившимся амилоидозом клинические проявления сохраняются после трансплантации.
При запястном туннельном синдроме применяется хирургическая декомпрессия срединного нерва. Для уменьшения боли в суставах используются НПВП.
Японскими авторами опубликованы данные об эффективности лазерной деструкции амилоидных фибрилл бета2-микроглобулина. Метод предлагается как для предотвращения отложения амилоида, так и для разрушения имеющихся депозитов.
Прогноз жизни при амилоидозе зависит от характера расположения амилоидных депозитов и степени повреждения органов-мишеней. В любом случае в отсутствие лечения заболевание неуклонно прогрессирует. При естественном течении AL- амилоидоза продолжительность жизни составляет в среднем 1-2 года. Наиболее низкая выживаемость ассоциирована с развитием застойной сердечной недостаточности (6 мес) и синдрома мальабсорбции (7,7 мес), при периферической полинейропатии — 56 месяцев, при нефротическом синдроме — 17 месяцев (данные клиники Мэйо). Прогноз при АА-амилоидозе зависит от природы основного заболевания и возможности его контроля. Своевременная и активная терапия воспалительного заболевания способна замедлить прогрессирование амилоидоза, а иногда уменьшить отложения амилоида.
источник
1. Осложнения системного AL-амилоидоза. Рестриктивная кардиомиопатия — характерный симптом, наблюдаемый у одной трети пациентов и приводящий к летальному исходу в половине случаев. Часто встречается вовлечение в патологический процесс почек с протеинурией и прогрессирующей почечной недостаточностью. Поражения кишечника могут вызывать нарушения моторики (часто вследствие вегетативной нейропатии), мальабсорбцию, перфорацию, кровотечение или непроходмость.
Периферическая нейропатия встречается в одной пятой случаев и обычно проявляется болезненной чувствительной полинейропатией с последующим развитием моторных нарушений. Вегетативная нейропатия, приводящая к ортостатической гипотензии, импотенции и желудочно-кишечным расстройствам может встречаться изолированно или сочетано с периферической нейропатией.
2. Осложнения системного АА-амилоидоза. Прогрессирующая почечная недостаточность и нефротический синдром представляют собой типичные осложнения АА-амилоидоза. Хотя отложения амилоида в селезенке почти всегда наблюдаются при амилоидозе АА-типа, обычно они не имеют клинических симптомов. Поражение печени и вегетативной нервной системы хорошо описаны, но они обычно возникают на очень поздних стадиях заболевания. Амилоидоз сердца возникает менее чем в 2% случаев.
3. Осложнения наследственного амилоидоза. Семейная полинейропатия вызывает прогрессирующую периферическую и/или вегетативную нейропатию и кардиомиопатию, а в некоторых случаях — потерю зрения вследствие отложения амилоида в стекловидном теле. Аполипопротеин-А1-амилоидоз может быть причиной периферической нейропатии, но как и при аполипопротеин-А2, лизоцим и a-цепи фибриногена А вариантах, типичным является поражение почек или печени. Гелсолин-ассоциированный амилоидоз вызывает характерную клиническую картину решетчатой дистрофии роговицы и нейропатии черепных нервов.
4. Осложнения локализованного амилоидоза. Вероятны только локальные осложнения в виде зуда или кровотечения, редко — болезненность.
б) Прогноз и течение амилоидоза. Хотя последние достижения значительно увеличили среднюю продолжительность жизни, прогноз при системном амилоидозе остается неблагоприятным.
1. Прогноз AL-амилоидоза. Прогноз в особенности неблагоприятен при AL-амилоидозе, так как диагноз часто устанавливается слишком поздно, когда пациенты уже имеют выраженное поражение внутренних органов и/или нервной системы. Ранее опубликованы данные о 5-летней выживаемости, составляющей приблизительно 10%, и о 10-летней выживаемости — менее 5%, что не зависит от типа AL-амилоидоза, хотя последние исследования говорят о средней выживаемости 40 месяцев.
2. Прогноз АА-амилоидоза. Прогноз АА-амилоидоза зависит от того, насколько выражено нарушение функции почек, и может ли лежащее в основе хроническое воспалительное заболевание подвергаться успешной супрессии, в той степени, чтобы уровни SAA в плазме оставались в нормальных пределах. В когортном исследовании 374 пациентов средняя выживаемость была чуть более 11 лет, у трети из них развилась терминальная почечная недостаточность, что явилось основной причиной смерти.
3. Прогноз наследственного амилоидоза. При семейной амилоидной полинейропатии (FAP) без трансплантации печени средняя выживаемость составляет 10-15 лет. Смерть наступает от прогрессирующей нейропатии и/или кардиомиопатии. При других формах наследственного амилоидоза, причиной смерти является либо почечная, либо печеночная недостаточность, но период до наступления терминальной недостаточности органа сильно варьирует.
4. Прогноз локализованного амилоидоза кожи. Варианты амилоидоза, ограниченные поражением кожи, обычно не приводят к смертельному исходу.
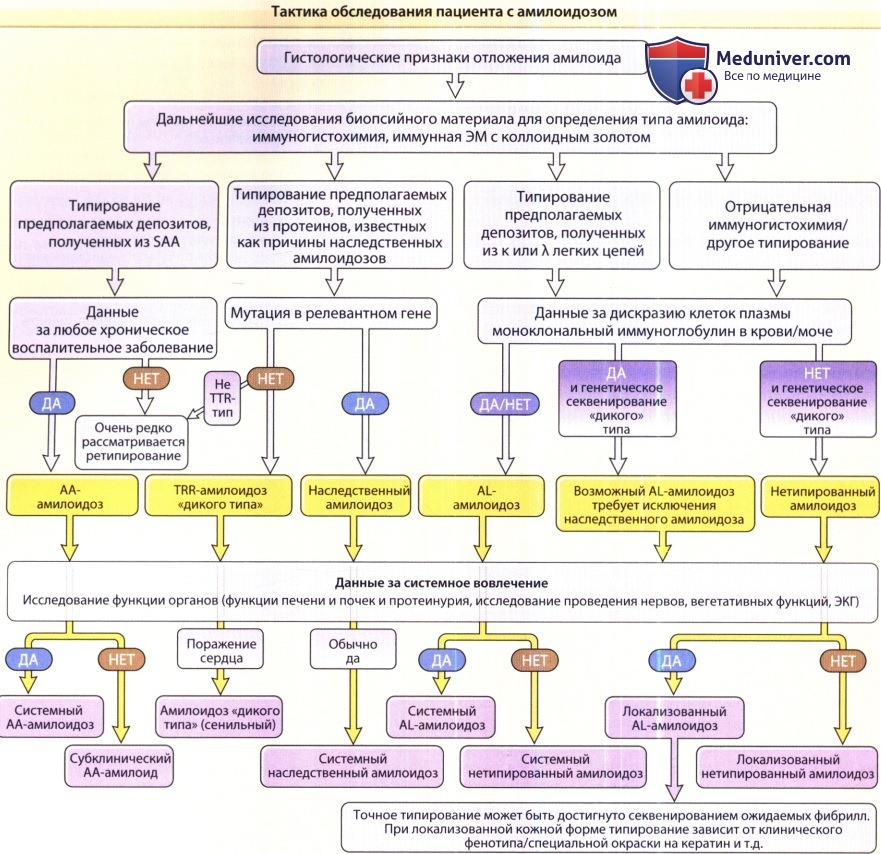 Тактика обследования пациента с амилоидозом.
Тактика обследования пациента с амилоидозом.
Ведение больного полностью зависит от точного типирования. Это является как высоко специализированной, так и комплексной задачей.
Точная установка типа амилоидоза предполагает ряд специальных тестов и лучше всего проводится в центрах, имеющих опыт таких исследований.
КМ — костный мозг; ЭКГ—электрокардиография; ЭМ — электронная микроскопия;
SAA—сывороточный протеин амилоид A; SAP — сывороточный компонент амилоид P; TTR — транстиретин.
— Рекомендуем далее ознакомиться со статьей «Современное лечение амилоидоза»
Редактор: Искандер Милевски. Дата публикации: 2.11.2018
источник
А милоидоз — общее определение для группы заболеваний, которых объединяет отложение в различных тканях и органах белка амилоида. На сегодня известно больше 20 предшественников амилоида, которые при благоприятных обстоятельствах депонируются в тех или иных частях тела.
Амилоид имеет фибриллярную структуру, которая была определена при изучении белка под электронным микроскопом.
Для амилоидоза сердца характерно быстрое прогрессирование, поэтому лечение является наиболее эффективным на раннем этапе развития болезни. Если же адекватное терапия не проводится, тогда за один месяц стенка сердца может утолщаться на 1,5 мм и более, а это грозит очень быстрой сердечной недостаточностью и смертью (примерно через 6 мес).
Видео: Что такое амилоидоз, чем он опасен, как с ним бороться?
Термин “амилоидоз” относится не к одной патологии, а к совокупности заболеваний, при которых инфильтрат на основе белка откладывается в тканях как бета-плиссированные листы. Подтип заболевания определяется тем, какой белок осаждается, хотя описаны десятки подтипов, большинство из них невероятно редки или имеют тривиальное значение.
Обозначения
В правильной номенклатуре используется буква “А” для амилоида, за которой следуют буква(ы), относящиеся к основному депонированному белку. Например, амилоидоз легкой цепи представляет собой “AL” (“А” для амилоида и “L” для легкой цепи). Амилоидоз транстиретина определяется как “ATTR” (“A” для амилоида и “TTR” для транстиретина). Такие термины, как “первичный амилоидоз”, “вторичный амилоидоз”, “старческий амилоидоз” и “семейная амилоидная кардиомиопатия”, часто приводят к путанице, поэтому их лучше всего избегать.
В подавляющем большинстве сердечный амилоидоз вызывается одним из двух белков: легкими цепями или транстиретином. Клинические проявления и лечение этих двух подтипов между собой отличаются.

Является наиболее часто диагностируемой формой системного амилоидоза. Также раньше определялся как “первичный амилоидоз”, но по приведенным выше причинам это названием не будет использоваться.
Плазменные клетки находятся в основном в костном мозге и продуцируют большое количество антител. Антитела состоят из тяжелых цепей и легких цепей. Когда плазматическая клетка становится клоновой (по существу, превращается в злокачественную), она и ее клоны обычно продуцируют клональное антитело и клональный избыток легкой цепи, связанный с этим антителом. В этом процессе возможно три возможных исхода:
- Клон плазменной клетки берет на себя небольшую часть костного мозга, и легкая цепь безвредно выводится с мочой. Это состояние называется моноклональной гаммапатией неопределенного значения.
- Клон плазменной клетки поглощает большую часть костного мозга, что потенциально приводит к гиперкальциемии, анемии, литическим поражениям и / или почечной дисфункции. Это состояние называется миеломой.
- Клон плазменной клетки продуцирует легкую цепь, которая склонна к неправильной замене на бета-плиссированные листы (β-складчатый слой). Эти легкие цепи циркулируют в кровотоке и осаждаются в одной или нескольких тканях. Это состояние называется AL-амилоидозом.
AL-амилоидные отложения могут встречаться практически в любой ткани, а характер участия органа варьируется от пациента к пациенту (таблица 1).
Распространенные внесердачные участки поражения и связанные с ними проявления следующие:
- почки (альбуминурия и потенциальная почечная недостаточность);
- печень (повышение щелочной фосфатазы и потенциальная печеночная недостаточность);
- желудочно-кишечный тракт (дисфагия, запор, мальабсорбция и кровотечение);
- язык (макроглоссия);
- нервная система (периферическая невропатия и вегетативная дисфункция).
В крайне редких случаях у одного больного определяется клиническое участие всех этих органов и систем.
Таблица 1: Амилоидные подтипы и клинические характеристики
| Подтип | Демография | Вовлечение органа | Гипертрофия левого желудочка | Терапия |
| AL | M ≈ Ж Возраст 40-80 | Любой (сердце, почка, ГМ, язык, нервы, печень, мягкие ткани) | + | Химиотерапия или трансплантация стволовых клеток |
| Дикий тип (wild type) ATTR M | >>> Ж Возраст 65-95 | Сердце (синдром кистевого туннеля) | +++ | Поддерживающая терапия, ведутся клинические испытания |
| Мутантный АТТR | M >> Ж Возраст 55-75 | Сердце и нервы (синдром кистевого туннеля) | +++ | Поддерживающая терапия, ведутся клинические испытания |
Сердечные проявления при амилоидозе следующие:
- Сердечная недостаточность (СН), которая может быть диастолической и систолической.
- Аритмии (тахиаритмии / брадиаритмии).
Основным признаком является желудочковая гипертрофия, наблюдаемая при эхокардиографии с очень низким электрическим напряжением электрокардиограммы (ЭКГ).
Натрийуретические пептиды обычно повышаются при амилоидозе сердца, а анализы тропонина часто хронически положительны при низких уровнях (0,1-1 нг / мл) из-за продолжающегося постепенного разрушения кардиомиоцитов.
Транстиретин (преальбумин) представляет собой белок, продуцируемый печенью, который функционирует как транспортер тироксина и ретинола. Он преимущественно циркулирует как гомотетрамер с небольшим количеством транстиретина, находящегося в мономерной форме. Эта форма транстиретина подвержена неправильной замене и постепенно откладывается в виде амилоидных отложений.
Существуют два основных подтипа АТТR амилоидоза:
- ATTR дикого типа
- Мутантный ATTR.
В случае АТТR дикого типа белок транстиретина является нормальным (немутированным). В течение десятилетий он постепенно откладывается в виде амилоидных накоплений. Хотя небольшое количество отложений может возникать в мягких тканях (вызывающих синдром кистевого туннеля) и сосудистой сети, первичные патологические отложения определяются в сердце.
Мутантный тип ATTR амилоидоза сердца является наследственным заболеванием. При нем определяются патологические мутации в транстиретиновом гене, что приводит к ускоренному отложению амилоида. Мутантный АТТR наиболее часто поражает и нервные волокна, причем картина осаждения в значительной степени зависит от мутации.
Окончательный диагноз амилоидоза требует биопсии. Для проведения процедуры зачастую выбирается абдоминальная жировая прослойка, поскольку это место характеризуется легкой доступностью и низкой заболеваемостью. Однако этот участок тела обладает относительно низкой чувствительностью, поэтому даже в положительных случаях часто получаются неадекватные амилоидные отложения, свойственные для окончательного подтипа болезни (АТТR, АL и т. д.). Таким образом, практикуется биопсия органа, вовлеченного в патологический процесс (т. е. сердца при подозрении на сердечный амилоидоз).
При проведении эндомиокардиальной биопсии дается почти 100% достоверный результат о наличии или отсутствии амилоидоза сердца.
Определение подтипа амилоидоза сердца может быть выполнено иммунофлюоресценцией или образцы отправляются в лабораторию для проведения масс-спектрометрии. Если есть какие-либо сомнения относительно диагноза, основанного на окрашивании иммунофлюоресценцией, должна быть проведена масс-спектрометрия.
Дополнительные лабораторные тесты при AL-амилоидозе позволяют определить другую дисфункцию органа:
- протеинурию;
- концентрацию щелочной фосфатазы;
- количество циркулирующих легких цепей.
Анализы на концентрацию легких цепей могут быть полезны в качестве постановки предположительного диагноза до выполнения биопсии. В частности, нормальная величина циркулирующих легких цепей делают диагноз AL-амилоидоза маловероятным. Так подобные исследования необходимы для контроля реакции на химиотерапию.
При ATTR-амилоидозе проводится генетическое тестирование на определение гена транстиретина. Наличие патологической мутации может влиять на варианты клинических испытаний, прогнозирование участия органов и указывать на предположительное отношение к членам семьи.
Электрокардиография — важное исследование при любой форме сердечного амилоидоза. С ее помощью обнаруживается гипертрофия желудочков. Гипертрофия при амилоидозе представляет собой отложение амилоидных фибрилл, а не гипертрофию / гиперплазию миоцитов. Это объясняет сниженное напряжение ЭКГ, а не увеличенное, как это бывает при типичной гипертрофии.
Магнитно-резонансная томография сердца также проводится при подозрении на амилоидоз. В частности, может определяться глобальное трансмуральное или субэндокардиальное распределение сердечной ткани.
Эхокардиография — зачастую определяются деформации и базальные-, апикально-преобладающие нарушения.
Видео: Сердце при амилоидозе
Лечение AL-амилоидоза проводится согласно двум параллельным стратегиям:
- Устранение последствий дисфункции органа, то есть проводится симптоматическая терапия.
- Замедление прогрессирования заболевания, для чего убиваются клональные плазматические клетки (и, следовательно, уменьшается количество циркулирующих патологических легких цепей). Для этого используется химиотерапия.
Кардио-специфическое лечение
Практикуется при всех формах амилоидоза, с учетом ряда важных моментов:
- Делается акцент на регуляции объема жидкости в организме. Для этого применяются диуретики и ограничивается употребление соли.
- Дигоксин связывается с амилоидными фибриллами, что приводит к потенциальной интоксикации дигоксином даже при нормальных циркулирующих уровнях, поэтому этот препарат, как правило, избегают.
- При необходимости проводится контроль над аритмией. При этом нейрогормональные антагонисты, обычно используемые при СН, часто плохо переносятся и являются контрпродуктивными.
- Бета-блокаторы, ингибиторы ангиотензинпревращающего фермента и блокаторы рецепторов ангиотензина часто приводят к гипотонии (из-за вегетативной дисфункции и наличия небольшой полости левого желудочка с невозможностью увеличения объема).
- Дополнительно бета-блокаторы нередко усугубляют течение брадиаритмии.
Кардиостимуляторы необходимы при высокой распространенности болезни и снижении качества проводимости (особенно при ATTR-амилоидозе). Фибрилляция предсердий довольно распространена среди больных амилоидозом сердца и часто плохо переносится. Подобное состояние требует кардиоверсии и / или антиаритмической терапии (чаще всего амиодароном). Среди этой категории больных распространены желудочковые аритмии и внезапная сердечная смерть.
Исторически сложилось так, что имплантация кардиовертер-дефибрилляторов (КВД) не приобрела всеобщего признания из-за плохого прогноза, связанного с сердечным амилоидозом. Более свежие данные свидетельствуют о том, что КВД могут быть эффективной частью стратегии управления состоянием больных при амилоидозе сердца. В частности, имплантация устройства продлевает жизнь на 1 год и больше.
Трансплантация сердца
Поскольку сердечный амилоидоз является необратимым заболеванием и часто может быть связан с тяжелыми симптомами и высокими показателями смертности, трансплантация сердца рассматривается в качестве лечения отдельных пациентов. Дополнительно учитывается следующее:
- В лечении больных на AL-амилоидоз исключается возможность значительного участия других жизненно важных органов. При этом кардиальная трансплантация должна сопровождаться стратегией, основанной на химиотерапии для контроля производства легких цепей.
- При ATTR-амилоидозе должно быть исключено значительное неврологическое участие пациентов с семейными формами.
Во всех случаях рекомендуется проводить трансплантацию только в центрах, которые имеют практику по пересадке сердца пациентам с амилоидозом.
Химиотерапия
За последнее десятилетие возможности химиотерапии для лечения AL-амилоидоза заметно улучшились, так что у большинства больных достигается значительное сокращение (а иногда и полная нормализация) количества циркулирующих патологических легких цепей.
Химиотерапия обычно состоит из комбинации нескольких классов антинеопластов. В частности используется:
- алкилаторы (например, мелфалан или циклофосфамид);
- стероиды (например, дексаметазон);
- ингибиторы протеасомы (например, бортезомиб или карфилзомиб);
- иммуномодуляторы (например, леналидомид или помалидомид).
Альтернативная стратегия включает трансплантацию стволовых клеток (stem-cell transplant) на фоне высокодозной химиотерапии алкилирующим препаратом. Трансплантат со стволовыми клетками может быть эффективным методом снижения легких цепей, поэтому используется многими центрами.
Важно отметить, что единственное рандомизированное исследование, сравнивающее лечение стволовыми клетками со стандартной химиотерапией, показало, что трансплантат дает более низкие результаты. Также при использовании стандартных методов химиотерапии отмечаются более быстрые улучшения, поэтому пересадку стволовых клеток проводят редко, только в отдельных случаях.
Поскольку ATTR-амилоидоз не является злокачественным процессом, химиотерапия не играет настолько важную роль, как в случае с AL-амилоидозом. Несмотря на то, что в настоящее время не утверждены лекарства, модифицирующие болезнь, для лечения ATTR-амилоидоза были изучены некоторые препараты, часть из них находится на поздних стадиях клинической оценки. К ним относятся следующие:
- Дифлунизал (Diflunisal). Этот нестероидный противовоспалительный препарат, который одобрен для лечения артрита, стабилизирует тетрамерную форму транстиретина. Рандомизированное исследование продемонстрировало замедление прогрессирования заболевания среди пациентов с полинейропатией из-за мутантного ATTR-амилоидоза. Поскольку нестероидные противовоспалительные препараты относительно противопоказаны при СН, это лекарство вряд ли является хорошим вариантом при наличии амилоидной кардиомиопатии.
- Тафамидис(Tafam >Прогноз
Прогностическое заключение при амилоидозе зависит прежде всего от тяжести сердечного поражения. При AL-амилоидозе также имеет значение уровень циркулирующих легких цепей. Поскольку в последние годы возможности химиотерапии значительно расширились, прогноз при AL-амилоидоза заметно улучшился. Также средняя продолжительность жизни большинства пациентов, включая многих из тех, кто имеет значительное поражение сердца, уже измеряется не месяцами, как раньше, а годами.
Прогноз при ATTR-амилоидоз обычно лучше, чем при AL-амилоидозе, хотя обе формы заболевания по-прежнему характеризуются высокой годовой смертностью.
Для AL-амилоидоза были предложены различные системы составления прогностического заключения, причем основное внимание уделялось степени поражения сердца. Широко используемая система прогнозирования, опубликованная в 2004 году, основывалась исключительно на двух сердечных биомаркерах:
- тропонине (T или I);
- про-натрийуретической N-концевом пептиде В-типа.
Таким образом, постоянное улучшение схем химиотерапии с периода начатых изучений патологического состояния позволит повысить выживаемость больных на амилоидоз сердца.
- Отношение к амилоидозу сердца заметно изменилось за последнее десятилетие, при этом большее число больных было диагностировано, также заметно улучшились терапевтические возможности.
- Диагностические признаки амилоидоза сердца включают нетипичную гипертрофию желудочков с низким напряжением ЭКГ, необъяснимой СН и характерной дисфункцией других органов.
- Постановка окончательного диагноза основана на биопсии пораженного органа, к тому же это исследование имеет решающее значение для окончательного подтипирования амилоидных отложений (например, AL и ATTR).
- При AL-амилоидозе лечение должно начинаться незамедлительно с использованием принципов химиотерапии или трансплантации стволовых клеток.
- При ATTR-амилоидозе больные зачастую регистрируются для участия в одном из текущих клинических испытаний.
- Лечение сердца при амилоидозе в первую очередь основано на использовании диуретиков и аритмических препаратов.
- В отдельных случаях рассматривается возможность имплантации КВД или проведения трансплантации сердца.
Поскольку при амилоидозе сердца нередко требуется участие врачей различных специальностей, больному обычно рекомендуют обратиться в центр амилоидоза.
Видео: «Амилоиды — опасные белки внутри нас» Елена Венская
источник
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
По современным представлениям, лечение амилоидоза — это уменьшение количества белков-предшественников (или, если возможно, их удаление) с целью замедлить или приостановить прогрессирование амилоидоза. Неблагоприятный прогноз при естественном течении амилоидоза оправдывает применение некоторых агрессивных лекарственных режимов или других радикальных мер (химиотерапия высоких доз с последующей пересадкой аутологичных стволовых клеток у больных с AL-амилоидозом). Клиническое улучшение, которое может быть достигнуто с помощью этих видов лечения, заключается в стабилизации или восстановлении функции жизненно важных органов, а также в предотвращении дальнейшей генерализации процесса, что увеличивает продолжительность жизни больных. Морфологическим критерием эффективности лечения считают уменьшение отложений амилоида в тканях, что в настоящее время можно оценить, применяя радиоизотопную сцинтиграфию с сывороточным бета-компонентом. Кроме основных терапевтических режимов, лечение амилоидоза должно включать симптоматические методы, направленные на уменьшение выраженности застойной недостаточности кровообращения, аритмий, отёчного синдрома, коррекцию артериальной гипотензии или гипертензии.
 [1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [11], [12], [13]
[1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [11], [12], [13]
Цель лечения вторичного амилоидоза — подавление продукции белка-предшественника SAA, чего достигают лечением хронического воспаления, в том числе и хирургическим путём (секвестрэктомия при остеомиелите, удаление доли лёгкого при бронхоэктатической болезни), опухоли, туберкулёза. Особое значение в настоящее время придают лечению ревматоидного артрита, учитывая его лидирующее положение среди причин вторичного амилоидоза. При базисной терапии ревматоидного артрита цитостатическими препаратами: метотрексатом, циклофосфамидом, хлорамбуцилом, — назначаемыми на длительный срок (более 12 мес), амилоидоз развивается реже. У пациентов с уже развившимся амилоидозом лечение цитостатиками позволяет в большинстве случаев уменьшить клинические проявления амилоидной нефропатии. В результате лечения амилоидоза отмечают снижение протеинурии, купирование нефротического синдрома, стабилизацию функций почек. У части пациентов удаётся предотвратить развитие хронической почечной недостаточности или замедлить её прогрессирование, что существенно улучшает прогноз. Контроль эффективности лечения амилоидоза цитостатиками — нормализация концентрации С-реактивного белка в крови. Перспективным методом лечения, способным вытеснить традиционные цитостатики, считают применение ингибиторов ФНО-а.
Средство выбора для лечения амилоидоза АА при периодической болезни — колхицин. При его постоянном приёме можно полностью прекратить рецидивирование приступов у большинства больных и обеспечить профилактику развития амилоидоза. При развившемся амилоидозе длительный (возможно, пожизненный) приём колхицина в дозе 1,8-2 мг/сут приводит к ремиссии, выражающейся в ликвидации нефротического синдрома, уменьшении или исчезновении протеинурии у больных с нормальной функцией почек. При наличии хронической почечной недостаточности начальную дозу колхицина уменьшают в зависимости от величины клубочковой фильтрации, хотя в случае снижения концентрации креатинина в крови возможно повышение дозы до стандартной. Колхицин также предотвращает рецидив амилоидоза в трансплантированной почке. Больные хорошо переносят этот препарат. При диспепсии (наиболее частом побочном эффекте колхицина) нет необходимости в отмене средства: она, как правило, исчезает самостоятельно или при назначении ферментных препаратов. Пожизненный приём колхицина безопасен. Антиамилоидный эффект колхицина основан на его способности в эксперименте подавлять острофазовый синтез белка-предшественника SAA, блокировать образование амилоидускоряющего фактора, что тормозит образование фибрилл амилоида. Если эффективность колхицина при амилоидозе в рамках периодической болезни не вызывает сомнений, то имеются лишь единичные работы, свидетельствующие о его успешном применении у больных вторичным амилоидозом. Предположение о том, что препарат можно эффективно использовать для лечения АА-типа амилоидоза в целом, пока не доказано. Кроме колхицина, при АА-амилоидозе применяют диметил-сульфоксид, вызывающий резорбцию амилоидных отложений. Однако использование его в высоких дозах (не менее 10 г/сут), необходимых для успешного лечения, ограничено из-за крайне неприятного запаха, который исходит от больных при его приёме. Современным препаратом, направленным на резорбцию амилоида, является фибриллекс; его применение оправдано в качестве дополнения к основной терапии предрасполагающего заболевания или лечению колхицином.
При AL-типе амилоидоза, как и при миеломной болезни, цель лечения — подавление пролиферации или полная эрадикация клона плазматических клеток для уменьшения продукции лёгких цепей иммуноглобулинов. Этого достигают при назначении мелфалана в сочетании с преднизолоном. Лечение продолжают 12-24 мес 4-7-дневными курсами с интервалом 4-6 нед. Доза мелфалана составляет 0,15-0,25 мг/кг массы тела в сутки, преднизолона — 0,8 мг/кг массы тела в сутки. У больных с хронической почечной недостаточностью (СКФ менее 40 мл/мин) дозу мелфалана уменьшают на 50%. При наличии признаков прогрессирования амилоидоза через 3 мес лечения терапию необходимо прекратить. Несомненным показателем эффективности терапии через 12- 24 мес считают уменьшение протеинурии на 50% без нарушения функций почек, нормализацию повышенной до начала лечения концентрации креатинина в крови, исчезновение симптомов недостаточности кровообращения, а также уменьшение на 50% содержания моноклонального иммуноглобулина в крови и моче. Однако длительное (не менее 12 мес) лечение можно провести не у всех больных, поскольку прогрессирование болезни может опережать положительный эффект мелфалана: он обладает миелотоксическими свойствами, что может привести к развитию лейкемии или миелодисплазии. Лечение амилоидоза мелфаланом и преднизолоном по указанной схеме позволяет избежать миелотоксичности мелфалана: положительного эффекта достигают у 18% больных, причём лучшие результаты отмечают при НС без нарушения функций почек и недостаточности кровообращения. Продолжительность жизни пациентов, у которых развился положительный ответ на лечение, составляет в среднем 89 мес.
В последнее время при AL-амилоидозе (не только в рамках миеломной болезни, но и при первичном амилоидозе) всё чаще применяют более агрессивные схемы полихимиотерапии с включением винкристина, доксорубицина, циклофосфана, мелфалана, дексаметазона в разных комбинациях. Последние исследования свидетельствуют о большей эффективности химиотерапии высоких доз. Так, R.L. Comenzo и соавт. в 1996 г. опубликовали предварительные результаты лечения 5 больных AL-амилоидозом внутривенными вливаниями мелфалана в дозе 200 мг/м 2 поверхности тела с последующим введением аутологичных стволовых клеток (CD34 + ) крови. Аутологичные стволовые клетки получают методом лейкафереза крови больного после предварительной их мобилизации из костного мозга под влиянием введённого извне гранулоцитарного колониестимулирующего фактора. Однако тяжёлый агранулоцитоз и другие осложнения этой терапии существенно ограничивают применение терапии сверхвысокими дозами мелфалана, в частности, у больных с недостаточностью кровообращения. Низкие показатели выживаемости больных AL-амилоидозом не позволяют с определённостью оценить эффективность этих схем. Применение колхицина для лечения AL-типа амилоидоза оказалось неэффективным.
Цель лечения — уменьшение количества белка-предшественника путём увеличения клиренса бета2-микроглобулина при проведении современных методов очищения крови: высокопоточного гемодиализа на синтетических мембранах, позволяющего улучшить абсорбцию р\,-микроглобулина, гемофильтрации, иммуносорбции. При этих методах можно снизить концентрацию белка-предшественника примерно на 33%, что позволяет отсрочить или затормозить развитие диализного амилоидоза. Однако единственным по-настоящему эффективным способом лечения остаётся трансплантация почки. После неё содержание бета2-микроглобулина снижается до нормальных значений, что сопровождается быстрым исчезновением клинических признаков амилоидоза, хотя отложения амилоида в костях сохраняются долгие годы. Редукция симптомов заболевания, по-видимому, связана с противовоспалительным действием иммуносупрессивной терапии после трансплантации и, в меньшей степени, с прекращением процедур гемодиализа.
Средство выбора лечения ATTR-типа амилоидоза — трансплантация печени, при которой удаётся удалить источник синтеза амилоидогенного предшественника. После этой операции, если отсутствуют признаки далеко зашедшей невропатии, пациента можно считать практически излеченным.
Поскольку хроническая почечная недостаточность — одна из основных причин смерти больных системным амилоидозом, проведение гемодиализа или постоянного амбулаторного перитонеального диализа позволяет улучшить прогноз этих пациентов. Выживаемость больных амилоидозом при проведении гемодиализа, независимо от его типа, сопоставима с выживаемостью больных другими системными заболеваниями и сахарным диабетом. При этом хорошую и удовлетворительную реабилитацию отмечают у 60% пациентов с АА- и AL-типами болезни. Поражение сердца и сосудов бывает основной причиной смерти больных амилоидозом при проведении гемодиализа. Постоянный амбулаторный ПД имеет некоторые преимущества перед гемодиализом, поскольку нет необходимости в постоянном сосудистом доступе, не возникает артериальной гипотензии во время процедуры диализа, а у больных с AL-типом амилоидоза во время процедуры возможно удаление лёгких цепей иммуноглобулинов. Трансплантация почки одинаково эффективна при обоих типах системного амилоидоза. Пятилетняя выживаемость больных и трансплантата составляют 65 и 62% соответственно и сопоставима с соответствующими показателями в других группах больных с хронической почечной недостаточностью.
Трансплантация почки показана больным с медленным прогрессированием амилоидоза без поражения сердца и ЖКТ. Амилоидоз в трансплантированной почке возникает, по разным данным, примерно у 30% больных, однако он становится причиной потери трансплантата всего у 2-3% пациентов.
источник
«Амилоидоз» — термин, объединяющий группу заболеваний, которые отличаются большим разнообразием клинических проявлений и характеризуются внеклеточным отложением нерастворимых патологических фибриллярных белков в органах и тканях. Впервые эта патология бы
«Амилоидоз» — термин, объединяющий группу заболеваний, которые отличаются большим разнообразием клинических проявлений и характеризуются внеклеточным отложением нерастворимых патологических фибриллярных белков в органах и тканях. Впервые эта патология была описана в XVII в. Боне — саговая селезенка у больного с абсцессом печени. В середине XIX в. Вирхов применил ботанический термин «амилоид» (от греч. amylon — крахмал) для описания внеклеточного материала, обнаруженного в печени при аутопсии, так как полагал, что он близок по структуре к крахмалу. Впоследствии была установлена белковая природа отложений, однако термин «амилоид» сохранился до настоящего времени.
В 20-е гг. XX столетия Бенхольд предложил окрашивать амилоид конго-красным, затем был обнаружен эффект двойного лучепреломления в поляризованном свете — изменение кирпично-красной окраски на яблочно-зеленую. В 1959 г. Коген и Калкинс с помощью электронной микроскопии установили фибриллярную структуру амилоида.
Эволюцию претерпели и клинические представления об амилоидозе: Рокитанский в 1842 г. установил связь «сальной болезни» с туберкулезом, сифилисом, риккетсиозами; Уилкс в 1856 г. описал «жирные органы» у больного, не имевшего никаких сопутствующих заболеваний; Аткинсон в 1937 г. обнаружил амилоидоз у пациентов с миеломной болезнью. Выделены были старческие (Сойка, 1876) и наследственные (Андраде, 1952) формы заболевания, амилоидоз разделяли на генетический, первичный и вторичный, и, наконец, в 1993 г. была принята классификация ВОЗ, построенная на специфичности основного фибриллярного белка амилоида.
В нашей стране большой вклад в развитие представлений об амилоидозе внесли Е. М. Тареев, И. Е. Тареева, В. В. Серов. Огромная роль в изучении первичного и генетических вариантов амилоидоза и периодической болезни принадлежит О. М. Виноградовой, чьи монографии, изданные в 1973 и 1980 гг., не утратили своей актуальности и в наши дни.
В настоящее время амилоидоз принято клинически разделять на системные и локальные формы. Среди системных форм, в зависимости от состава фибриллярных отложений, выделяют четыре типа (табл. 1).
К локальным формам амилоидоза в настоящее время относят болезнь Альцгеймера (A-бета, фибриллы состоят из β-протеина, откладывающегося в головном мозге), амилоидоз островков поджелудочной железы, возможно, имеющий патогенетическую связь с диабетом 2 типа, амилоидоз, возникающий в эндокринных опухолях, амилоидные опухоли кожи, назофарингеальной области, мочевого пузыря и другие редкие виды.
Развитие AL-амилоидоза возможно при миеломной болезни, болезни Вальденстрема, В-клеточных лимфомах, и оно может быть идиопатическим при первичном амилоидозе. Все эти варианты объединены общим патогенезом, первичный амилоидоз представляет наибольшую трудность для распознавания в связи с отсутствием явных признаков гематологического заболевания, поэтому именно на данной форме стоит остановиться подробно.
При первичном амилоидозе, доброкачественной плазмоклеточной дискразии, родственной множественной миеломе, аномальные клоны плазматических клеток костного мозга продуцируют амилоидогенные иммуноглобулины. Некоторые аминокислоты в вариабельных участках легких цепей этих иммуноглобулинов занимают необычную позицию, что приводит к их нестабильности и обусловливает склонность к фибриллогенезу. У больных с первичным амилоидозом содержание плазматических клеток в костном мозге повышено до 5—10% (в норме их менее 4%, при миеломной болезни — более 12%), и они продуцируют определенный изотип легких цепей иммуноглобулинов, преобладающий при иммуногистохимическом окрашивании. Свободные моноклональные легкие цепи преобладающего лямбда- или (реже) каппа-изотипа определяются в крови и в моче, но содержание их ниже, чем при миеломной болезни.
Клиническая картина первичного амилоидоза многообразна и определяется преимущественным вовлечением в патологический процесс тех или иных органов — сердца, почек, нервной системы, желудочно-кишечного тракта, печени и др. Первыми симптомами являются слабость и потеря веса, но на этой стадии, до появления органных симптомов, диагноз устанавливается крайне редко.
Органами-мишенями при AL-амилоидозе чаще всего становятся почки и сердце. Поражение почек проявляется нефротическим синдромом, персистирующим и при наступлении ХПН, гематурия и артериальная гипертензия не характерны.
При отложении амилоида в миокарде развиваются разнообразные нарушения ритма, прогрессирующая сердечная недостаточность, чему могут предшествовать бессимптомные изменения на ЭКГ в виде снижения вольтажа зубцов. Эхокардиографическое исследование выявляет концентрическое утолщение стенок левого и правого желудочков, уменьшение объема полостей сердца, умеренное снижение фракции выброса, диастолическую дисфункцию миокарда левого желудочка.
Часто отмечаются симптомы вовлечения нервной системы — вегетативной, в виде ортостатической гипотензии, и периферической — в виде расстройств чувствительности. В последние годы стали описывать также поражения ЦНС, хотя ранее считалось, что они не характерны для первичного амилоидоза.
Диспептические явления (ощущение переполнения, запоры, поносы) и синдром нарушенного всасывания могут быть обусловлены как поражением вегетативной нервной системы, так и амилоидозом желудочно-кишечного тракта. Очень характерна гепатомегалия, природу которой следует дифференцировать между застойными явлениями вследствие сердечной недостаточности и амилоидным поражением печени. Последнее подтверждается повышением уровня щелочной фосфатазы сыворотки крови. Селезенка поражается часто, однако спленомегалия обнаруживается не всегда и большого клинического значения не имеет.
Макроглоссия, классический признак первичного амилоидоза, отмечается у 20% пациентов, инфильтрация мягких тканей может приводить к атрофии мышц, кожи, дистрофии ногтей, алопеции и появлению опухолевидных образований — амилоидом.
Реже встречается поражение сосудов, симптомами которого являются периорбитальная пурпура — «глаза енота» и экхимозы. Могут наблюдаться кровотечения, в том числе мочепузырные, обусловленные как изменением сосудистой стенки, так и нарушением свертывающей системы, в первую очередь дефицитом X-фактора, который связывается с амилоидом. Дефицитом факторов свертывания принято объяснять и характерный для амилоидоза тромбоцитоз.
Амилоидоз легких часто обнаруживается лишь при аутопсии. Однако в некоторых случаях одышка, кровохарканье и гидроторакс могут быть обусловлены не только застойной сердечной недостаточностью и нефротическим синдромом, но и амилоидным поражением легких. Возможны отложение амилоида в альвеолах и развитие легочных амилоидом. Рентгенологически могут выявляться сетчатые и нодулярные изменения в легочной ткани.
Поражение надпочечников может привести к надпочечниковой недостаточности, нередко остающейся нераспознанной, так как гипотензия и гипонатриемия рассматриваются как симптомы сердечной недостаточности и поражения вегетативной нервной системы. У 10—20% больных может иметь место гипотиреоз как проявление поражения щитовидной железы, нередко встречается увеличение подчелюстных слюнных желез.
Диагноз первичного амилоидоза помимо указанных клинических черт, которые могут быть сходными и при вторичном амилоидозе, базируется на ряде лабораторных данных. У 85% пациентов при иммуноэлектрофорезе белков сыворотки крови и мочи выявляются моноклональные иммуноглобулины. При рутинных исследованиях те же моноклональные иммуноглобулины обнаруживаются в моче в виде белка Бенс-Джонса. Биопсия костного мозга позволяет провести дифференциальный диагноз с множественной миеломой, а также выявить умеренное повышение количества плазматических клеток и их моноклональность при иммуногистохимическом окрашивании.
Однако даже сочетания характерной клинической картины и наличия моноклональных плазмоцитов и белков еще недостаточно для подтверждения диагноза первичного амилоидоза. Решающую роль здесь играют данные биопсии. Наименее инвазивной является аспирация подкожной жировой клетчатки передней брюшной стенки, дающая 80—90% положительных результатов при AL-амилоидозе (в нашей стране этот метод пока не нашел применения). Определенное диагностическое значение имеет биопсия десны и слизистой оболочки прямой кишки, но процент положительных результатов широко варьирует, в зависимости от стадии процесса, поэтому целесообразно выполнение биопсии одного из пораженных орга-нов — почки, печени, сердца, дающее почти 100% положительных результатов при амилоидозе AL-типа.
В первую очередь биопсийный материал окрашивается конго-красным. При обнаружении конгофилии исследуемого материала необходимо его исследование в поляризованном свете, эффект двойного лучепреломления характерен только для амилоида, другие конгофильные вещества яблочно-зеленой окраски не приобретают. После этого желательно типирование амилоида. Наиболее точным является иммуногистохимический метод с использованием моноклональных антител к белкам-предшественникам амилоида. Однако в настоящее время в нашей стране он практически недоступен. Поэтому для диагностики используется окраска с помощью растворов щелочного гуанидина или перманганата калия, что позволяет, хотя и косвенно, определить тип фибриллярных отложений.
Прогноз при первичном амилоидозе хуже, чем при других формах заболевания, средняя продолжительность жизни не превышает двух лет, при наличии поражения сердца или мультисистемного поражения без лечения больные погибают в течение нескольких месяцев. Наиболее частыми причинами смерти являются сердечная и почечная недостаточность, сепсис, сосудистые осложнения и кахексия. Патогенетическое сходство с миеломной болезнью позволяет рассчитывать на торможение прогрессирования заболевания при химиотерапии, проводимой с целью подавления моноклональных плазмоцитов. Существует несколько схем лечения (табл. 2).
Применение химиотерапии в случае успеха лечения позволяет увеличить продолжительность жизни больных на срок от 10 до 18 мес. Но эффективность терапии невысока, в частности, в связи с тем, что во многих случаях прогрессирование заболевания приводит к гибели больных до завершения курса лечения, а также из-за развития цитопении, инфекционных осложнений, фатальных нарушений ритма при лечении сверхвысокими дозами дексазона. Применение высоких доз мельфолана с трансплантацией аутологичных стволовых клеток позволяет достичь ремиссии более чем в 50% случаев, однако использование этого метода ограничено тяжестью состояния, возрастом больных, функциональными нарушениями со стороны сердца и почек. Во многих случаях возможна лишь симптоматическая поддерживающая терапия.
Развитие AA-амилоидоза происходит при хронических воспалительных процессах, предшественниками AA-амилоида являются сывороточные острофазовые белки, α-глобулины, продуцируемые клетками разных типов, в основном нейтрофилами и фибробластами. Вторичный амилоидоз развивается при ревматоидном артрите, болезни Бехтерева, псориатическом артрите, различных опухолях, лимфогранулематозе, неспецифическом язвенном колите и болезни Крона, при периодической болезни (семейной средиземноморской лихорадке), а также при туберкулезе, остеомиелите, бронхоэктатической болезни.
Характерными клиническими особенностями АА-амилоидоза является поражение почек у большинства пациентов, а также относительно редкое поражение печени и/или селезенки (около 10%) и сердца (выявляется лишь при эхокардиографии). Макроглоссия для вторичного амилоидоза не характерна. Диагноз основан на сочетании амилоидоза почек и хронического воспалительного заболевания, подтверждением служит иммуногистохимическое окрашивание биопсийного материала, в нашей стране используются уже упомянутые выше косвенные окрасочные методы.
Прогноз во многом зависит от природы основного заболевания, при естественном течении у трети больных через 5 лет от момента выявления протеинурии развивается почечная недостаточность. При периодической болезни пятилетняя выживаемость составляет 25%.
Лечение основано на подавлении очага — источника продукции сывороточных белков-предшественников. Удаление опухолей, секвестрэктомия, резекция кишки, лечение туберкулеза, уменьшение активности ревматоидного артрита (при использовании цитостатиков) приводят к прекращению прогрессирования амилоидоза, а иногда и к обратному развитию клинических проявлений, в частности нефротического синдрома.
Применение колхицина при периодической болезни является методом выбора, эффективность его доказана, лечение предотвращает развитие амилоидоза и тормозит его прогрессирование. При других формах вторичного амилоидоза эффективность колхицина не подтверждена.
Сенильные и наследственные формы системного амилоидоза, так же как и локальные формы, встречаются редко, диализный амилоидоз хорошо известен специалистам, в общей практике с ним сталкиваться практически не приходится.
Симптоматическая терапия зависит не от типа амилоидоза, а от пораженных органов-мишеней (табл. 3).
Амилоидоз, особенно первичный, считается нечастой патологией, однако в действительности он не столько редко встречается, сколько с трудом диагностируется. Адекватная диагностика требует не только знания клиники и патогенеза данного заболевания, но и наличия определенных диагностических возможностей. Чтобы проиллюстрировать это положение, приведем собственные данные (см. таблицу 4). В нефрологическом отделении МГКБ имени С. П. Боткина в 1993—2003 гг. наблюдалось 88 больных, которым был поставлен диагноз амилоидоза.
Диагноз был подтвержден морфологически у всех больных с AL-амилоидозом, старческим и неуточненным по типу амилоидозом, и у 30 пациентов со вторичным амилоидозом — всего в 53 случаях. У 12 больных выполнялась биопсия почки, а у двоих — биопсия печени, у восьми — биопсия кишки, в 12 случаях — десны, еще в 19 случаях диагноз был подтвержден при морфологическом исследовании секционного материала.
В большинстве случаев диагноз амилоидоза был установлен впервые в результате обследования в нефрологическом отделении. Нами было проведено сопоставление среди больных с AL-амилоидозом направительного и клинического диагнозов (табл. 5).
Лишь в двух случаях из 20 (10%) направительным диагнозом был «первичный амилоидоз», причем одному из этих больных он был поставлен в клинике терапии и профзаболеваний ММА, а другому — в зарубежной клинике.
Все больные, у которых диагностировалась миеломная болезнь с развитием AL-амилоидоза, были переведены в гематологические отделения. Из 11 больных с первичным амилоидозом семь пациентов получали химиотерапию комбинацией мельфолана с преднизолоном внутрь прерывистыми курсами, четверо из них — в сочетании с диализным лечением, и еще одна больная — только диализное и симптоматическое лечение. Из числа этих больных пять человек умерли в сроки от двух недель до двух лет от начала лечения (все с почечной недостаточностью и полиорганным поражением), один больной находится на диализе, один больной был направлен на трансплантацию аутологичных стволовых клеток, и одна больная получает лечение до настоящего времени. У одного пациента химиотерапия отложена в связи с наличием длительно не рубцующейся язвы желудка, и еще двое больных отказались от лечения.
Среди больных с вторичным амилоидозом в нашем исследовании преобладали пациенты с ревматоидным артритом, на втором месте среди причин — хронический остеомиелит и псориатический артрит, остальные заболевания встречались реже (табл. 6).
Лечение ревматоидного артрита и псориатического артрита проводилось с применением цитостатиков (метатрексата, азатиоприна), хотя во многих случаях возможности терапии были ограничены из-за наличия ХПН и сопутствующей патологии. Больные с хроническим остеомиелитом были направлены в отделения гнойной хирургии. Пациенты с болезнью Бехтерева и болезнью Крона получали специфическое лечение, больные с ХНЗЛ и туберкулезом также были направлены в профильные стационары. Одна из больных с опухолью желудка была успешно оперирована, и на протяжение четырех лет наблюдения нефротический синдром постепенно регрессировал, в остальных случаях опухолей распространенность процесса позволяла проводить только симптоматическую терапию, больной с лимфогранулематозом поступил в терминальном состоянии. Смертность среди пациентов со вторичным амилоидозом составила 38% (за счет больных с далеко зашедшим поражением на момент постановки диагноза). Все больные с периодической болезнью получали терапию колхицином.
Особенности диагностики и применения современных методов лечения первичного амилоидоза можно проиллюстрировать на следующем примере: больная К., 46 лет, впервые госпитализирована в конце октября 2002 г. с жалобами на отеки на ногах, сердцебиения, аменорею. В анамнезе — простудные заболевания, аппендэктомия, два нормальных срочных родоразрешения, указаний на заболевание почек, какие-либо хронические заболевания нет. В апреле 2002 г. перенесла острую пневмонию в верхней доле правого легкого, лечилась амбулаторно, получала инъекции абактала, линкомицина. В связи с локализацией пневмонии была обследована в туберкулезном диспансере, диагноз туберкулеза исключен. В начале июня впервые появились отеки на ногах, по поводу которых не обследовалась. Отеки через короткое время самостоятельно ликвидировались, затем возобновились. Больная была госпитализирована в терапевтический стационар, при обследовании выявлена протеинурия до 1,65%, гипопротеинемия (общий белок сыворотки крови 52 г/л), артериальное давление в норме (120/80 мм рт. ст.), мочевой осадок без изменений, креатинин плазмы также в пределах нормы. Установлен диагноз «острый гломерулонефрит», проведено лечение ампициллином, курантилом, гепарином, триампуром, выполнена тонзиллэктомия. Протеинурия сохранялась, отеки постепенно нарастали, в связи с чем для дальнейшего обследования и лечения больная с диагнозом «хронический гломерулонефрит» была направлена в больницу им. С. П. Боткина.
При осмотре — кожа чистая, обычной окраски, анасарка, отеки массивные, плотные, определяется асцит, периферические лимфатические узлы не увеличены. АД 110/70 мм рт. ст., тоны сердца звучные, ясные, ритмичные, ЧСС 90 уд/мин, печень и селезенка не увеличены, диурез до 1000 мл/сут, стул регулярный, без патологических примесей. При обследовании выявлен нефротический синдром — протеинурия 3 г/л, мочевой осадок скудный, гиподиспротеинемия, гиперлипидемия (общий белок сыворотки крови 39 г/л, альбумины 12 г/л, глобулины 7-30-15-19% соответственно α1-α2-β-γ холестерин 17,8 ммоль/л, β-липопротеиды 250 ЕД), при анализе мочи на белок Бенс-Джонса — реакция отрицательная, суточная экскреция 17-КС не снижена. Клинический анализ крови и другие биохимические показатели в пределах нормы, коагулограмма — выраженная гиперфибриногенемия, повышение уровня РКФМ. Исследование иммуноглобулинов крови: Ig-A — 0,35, Ig-M — 35,7 (две нормы), Ig-G — 1,96 г/л. Рентгенография органов грудной клетки, костей черепа и таза, УЗИ брюшной полости, почек, щитовидной железы, ЭХО-КГ без патологии, УЗИ малого таза — признаки аденомиоза тела матки, ЭГДС — рефлюкс-эзофагит, хронический гастрит. При осмотре невропатологом патологии не найдено, онкологом установлена фиброзно-кистозная мастопатия.
С целью уточнения генеза нефротического синдрома под местной анестезией под УЗ-наведением выполнена тонкоигольная пункционная биопсия правой почки, осложнений не было. При исследовании биоптата в мезангии клубочков и во внегломерулярных сосудах отмечается отложение амилоида. Амилоид загружает до 25% сосудистых петель клубочков. При иммуногистохимическом исследовании специфической люминисценции не найдено. При обработке препаратов раствором щелочного гуанидина в течение 2 ч конгофилия и их свойства в поляризованном свете сохраняются, что характерно для AL-амилоидоза.
Для выяснения природы AL-амилоидоза выполнено иммунохимическое исследование крови и мочи в лаборатории «Иммунотест». Выявлена М-лямбда парапротеинемия со снижением уровня поликлональных иммуноглобулинов и парапротеинурия Бенс-Джонса лямбда-типа на фоне массивной неселективной протеинурии. Больная была консультирована гематологом, высказано предположение о наличии болезни Вальденстрема, произведена трепанобиопсия костного мозга. Заключение: в имеющихся костно-мозговых полостях видны клетки всех трех ростков нормального гемопоэза, а также лимфоидные клетки, не образующие скоплений. Диагноз болезни Вальденстрема отвергнут в связи с отсутствием лимфоидной инфильтрации костного мозга, увеличения лимфоузлов и селезенки и отсутствием субстрата опухоли.
Установлен диагноз первичного амилоидоза с поражением почек, нефротическим синдромом, сохранной почечной функцией, признаков иных органных поражений не выявлено. С января 2003 г. начата химиотерапия мельфоланом 16 мг/сут и преднизолоном 100 мг/сут, курсами по четыре дня каждые шесть недель. Проводится также симптоматическое лечение: фуросемид, верошпирон, препараты калия, фамотидин, переливания альбумина. К настоящему времени проведено пять курсов химиотерапии с хорошей переносимостью, отеки уменьшились, протеинурия снизилась до 1,8 г/л, несколько уменьшилась выраженность гиподиспротеинемии (общий белок 46 г/л, альбумины 18 г/л, α2-глобулины 20%). Функция почек остается сохранной, креатинин плазмы 1,3 мг/Дл, признаков поражения других органов и систем при контрольных динамических обследованиях не выявлено.
Данный случай наглядно иллюстрирует тот факт, что для диагностики амилоидоза необходимо морфологическое, иммунологическое и иммунохимическое обследование. Так, у нашей пациентки наиболее очевидным клиническим диагнозом был «хронический гломерулонефрит», и в отсутствии возможности выполнения биопсии почки именно этот диагноз, скорее всего, и был бы поставлен. Никаких клинических указаний на системный характер заболевания, хронический воспалительный процесс, заболевание системы крови, за исключением повышения уровня Ig-M, у больной не было. И лишь полученные при исследовании почечного биоптата данные повлекли за собой трепанобиопсию костного мозга и иммунохимическое исследование, что в совокупности позволило поставить диагноз первичного амилоидоза до появления системных повреждений. Патогенетическая терапия была начата хотя и на фоне уже развившегося нефротического синдрома, но до наступления почечной недостаточности и при загрузке лишь 25% клубочков амилоидом, что прогностически относительно благоприятно.
В заключение отметим, что амилоидоз представляет собой тяжелое заболевание с высоким уровнем летальности, которое чрезвычайно трудно диагностировать, однако своевременное и качественное обследование больных позволяет поставить диагноз в более ранние сроки, а своевременное назначение адекватной терапии, в свою очередь, дает возможность улучшить прогноз в этой группе больных.
- Варшавский В. А., Проскурнева Е. П. Значение и методы морфологической диагностики амилоидоза в современной медицине // Практическая нефрология. — 1998. — 2:16-23.
- Виноградова О. М. Первичный и генетический варианты амилоидоза. — М.: Медицина, 1980.
- Захарова Е. В., Хрыкина А. В., Проскурнева Е. П., Варшавский В. А. Случай первичного амилоидоза: трудности диагностики и лечения // Нефрология и диализ. — 2002. — 1:54-61.
- Рамеев В. В. Особенности поражения почек при AA и AL-амилоидозе: дисс . канд. мед. наук. — М., 2003.
- Козловская Л. В., Варшавский В. А., Чегаева Т. В. и др. Амилоидоз: современный взгляд на проблему // Практическая нефрология. — 1998. — 2:24-26.
- Rodney H., Raymond L.C and Skinner M. // The systemic Amyloidoses; New England Journal of Medicine, 1997. — 337:898-909.
- Dhodapkar M.V., Jagannath S., Vesole d. et al // Treatment of AL-amyloidosis with dexamethasone plus alpha interferon / Leuc Lymphoma. 1997. — 27(3-4):351-365
- Gertz M.A., Lacy M.Q., Lust J.A. et all // Phase II trial of high-dose dexamethasone for previosly treated immunoglobulin light-chain amyloidosis. Am J Hematol, 1999,61(2):115-119.
- Gertz M.A., Lacy M.,Q., Lust J.A. et al // Phase II trial of high-dose dexamethasone of untreated patients with primary systenic amyloidosis. Med Oncol 1999.- 16(2):104-109
- Sezer O., Schmid P.,Shweigert M. et al // Rapid reversal of nephrotic syndrome due to primary systemic AL amyloidosis after VAD and subsequent high-dose chemotherapy with autologous stem sell support. Bone Marrow Transplant. 1999. — 23(9): 967-969.
- Sezer O., Neimoller K., Jakob C. et al // Novel approaches to the treatment of primary amyloidosis. Expert Opin Investig Grugs. 2000. — 9(10):2343-2350
- Sezer O., Eucker J., Jakob C., Possinger K. // Diagnosis and treatment of AL amyloidosis. Clin Nephrol. 2000. — 53(6):417-423.
- Skinner M. «Amyloidosis» Current Therapy in Allergy, Immunology, and Rheumatology. Mosby-Year Book. 1996. — 235-240.
- Palladini G., Anesi E., Perfetti V. et al. A modified high-dose dexamethasone regimen for primary systemic (AL) amyloidosis. British Journal of Haematology. 2001. — 113:1044-1046.
Е. В. Захарова
Московская городская клиническая больница им. С. П. Боткина
источник