*Импакт фактор за 2017 г. по данным РИНЦ
Журнал входит в Перечень рецензируемых научных изданий ВАК.
Амилоидоз — группа заболеваний, характеризующихся отложением в органах и тканях особого белка фибриллярной структуры — амилоида. Клиническая картина амилоидоза многообразна, клиника зависит от вовлечения в патологический процесс того или иного органа, а определяющим в выборе лечебной тактики является установление причины его развития. В статье представлено клиническое наблюдение амилоидоза, ассоциированного с множественной миеломой. Особенностями данного клинического случая являются не верифицированная изначально нозологическая форма поражения сердца с ведущим синдромом сердечной недостаточности, рецидивирующее течение плеврита и отсутствие поражения почек в условиях доказанного амилоидоза. Диагностика вторичного AL-амилоидоза при множественной миеломе крайне затруднительна и основана на проведении гистологического и иммуногистохимического исследований костного мозга, секреции белка Бенс-Джонса λ. Наиболее эффективным методом лечения у пациентов молодого возраста являются протоколы терапии множественной миеломы (курсы VCD) для больных — кандидатов на аутологичную трансплантацию гемопоэтических стволовых клеток (ауто-ТГСК) с ее последующим выполнением.
Ключевые слова: AL-амилоидоз, множественная миелома, поражение сердца при амилоидозе, сердечная недостаточность.
Для цитирования: Загребнева А.И., Потешкина Н.Г., Кузнеченко Д.И., Бабак В.В. Системный амилоидоз, ассоциированный с множественной миеломой: клиническое наблюдение // РМЖ. 2018. №12(II). С. 107-109
Systemic amyloidosis associated with multiple myeloma: clinical observation
A.I. Zagrebneva 1,2 , N.G. Poteshkina 2 , D.I. Kuznechenko 1 , V.V. Babak 3
1 City Clinical Hospital, Moscow
2 Pirogov Russian National Research Medical University, Moscow
3 Nasonova Research Institute of Rheumatology, Moscow
An amyloidosis is a group of diseases characterized by the deposition in the organs and tissues of a particular protein of the amyloid fibrillar structure. The clinical picture of amyloidosis is diverse, the clinic depends on the involvement of a particular organ in the pathological process, and the determining factor in the choice of treatment tactics is to establish the cause of its development. The article presents a clinical case of amyloidosis associated with multiple myeloma. The features of this clinical case are presented as an initially uncertified nosological form of heart disease with the leading syndrome of cardiac insufficiency, a relapsing course of pleurisy and an absence of kidney damage under conditions of proven amyloidosis. Diagnosis of secondary AL-amyloidosis in multiple myeloma is challenging and is based on histological and immunohistochemical studies of the bone marrow, secretion of Bens-Jones λ-protein. The most effective treatment method in young patients is the protocols for the therapy of multiple myeloma (VCD courses) for patients-candidates for autologous transplantation of hematopoietic stem cells (auto-THSC) with its subsequent implementation.
Key words: AL-amyloidosis, multiple myeloma, heart disease in amyloidosis, cardiac insufficiency.
For citation: Zagrebneva A.I., Poteshkina N.G., Kuznechenko D.I., Babak V.V. Systemic amyloidosis associated with multiple myeloma: clinical observation // RMJ. 2018. № 12(II). P. 107–109.
В статье представлено клиническое наблюдение амилоидоза, ассоциированного с множественной миеломой. Особенностями данного клинического случая являются не верифицированная изначально нозологическая форма поражения сердца с ведущим синдромом сердечной недостаточности, рецидивирующее течение плеврита и отсутствие поражения почек в условиях доказанного амилоидоза.
Амилоидоз — группа заболеваний, для которых общим признаком является отложение в органах и тканях отсутствующего в норме особого белка фибриллярной структуры, названного амилоидом. Клиническая картина первичного амилоидоза многообразна и определяется преимущественным вовлечением в патологический процесс тех или иных органов — сердца, почек, нервной системы, желудочно-кишечного тракта, печени и др. Первыми симптомами являются слабость и потеря веса, но на этой стадии, до появления органных симптомов, диагноз устанавливается крайне редко.
Среди системных амилоидозов выделяют: АА, AL, ATTR, Ab2M-диализный амилоидоз. Одной из возможных причин развития AL-амилоидоза является множественная миелома, при которой аномальные клоны плазматических клеток костного мозга продуцируют амилоидогенные иммуноглобулины.
У больных с первичным амилоидозом содержание плазматических клеток в костном мозге повышено до 5–10% (в норме их менее 4%, при миеломной болезни — более 12%).
Плазматические клетки продуцируют определенный изотип легких цепей иммуноглобулинов, преобладающий при иммуногистохимическом окрашивании. Свободные моноклональные легкие цепи преобладающего лямбда- или (реже) каппа-изотипа определяются в крови и в моче, но содержание их ниже, чем при миеломной болезни.
Рассматриваемый клинический случай сопряжен с трудностью верификации как миеломной болезни (основной нозологии), так и сопряженного с ней амилоидоза.
Больная М., 50 лет, поступила с жалобами на общую слабость, резкое снижение толерантности к физическим нагрузкам, снижение веса на 10 кг за последний год. Кроме этого, отмечала резкое снижение диуреза, одышку при минимальной физической нагрузке, отеки нижних конечностей.
Из анамнеза: считает себя больной с 2011 г., когда перенесла ОРВИ, которая сопровождалась осиплостью голоса, вплоть до афонии. По данным обследования у оториноларингологов, а также при диагностической ларинго- и бронхоскопии патологии не выявлено. С конца декабря 2014 г. у пациентки появились отеки нижних конечностей, снижение толерантности к физическим нагрузкам (ранее вела активный образ жизни — посещение бассейна, еженедельные лыжные марафоны по 20 км в зимнее время).
В январе 2015 г. дебютировала одышка при минимальной физической нагрузке. По данным эхокардиографии (Эхо-КГ) заподозрена кардиомиопатия (обсуждалась гипертрофическая кардиомиопатия (ГКМП) без обструкции выносящего тракта). В это же время при рентгенографии органов грудной клетки выявлена внутригрудная лимфаденопатия. Выполнена торакотомия с биопсией средней доли легкого, лимфатических узлов средостения и плевры. Первичное морфологическое заключение: саркоидоз лимфатических узлов средостения. Проводилась терапия глюкокортикостероидами (ГКС) (триамцинолон 12 мг/сут). На фоне терапии в течение 1 мес. отмечалась отрицательная динамика: прогрессирование одышки, нарастание слабости, правостороннего гидроторакса до уровня IV ребра, выполнено дренирование плевральной полости ввиду его рецидивирующего течения. Увеличение дозы триамцинолона до 32 мг/сут оказалось неэффективным: сохранялся рецидивирующий плеврит справа, отмечалось усиление отеков нижних конечностей, одышки. ГКС были постепенно отменены.
Через год пациентке выполнена повторная торакоскопия с биопсией париетальной плевры, получено заключение: гранулематозное поражение плевры (очаговое скопление гранулематозных клеток в легких, продуктивный плеврит с гранулемами в плевре и атипией мезотелия). В дифференциальном ряду обсуждались саркоидоз и туберкулез. Атипичных клеток не выявлено. Результаты биопсии пересмотрены в НИИ пульмонологии, и убедительных данных за саркоидоз не получено. Туберкулез исключен фтизиатрами.
В связи с прогрессированием признаков и симптомов сердечной недостаточности и определением уровня NT-pro-BNP 380 пг/мл у пациентки с подозрением на ГКМП (по данным ранее проведенной Эхо-КГ), для верификации окончательного диагноза, выполнены коронароангиография (КАГ) и магнитно-резонансная томография (МРТ) сердца с контрастированием. По результатам КАГ исключен коронарный атеросклероз. Характер накопления контрастного препарата в области межжелудочковой перегородки, по данным МРТ сердца, мог соответствовать как воспалительным изменениям, так и гипертрофической кардиомио-
патии. Обращал на себя внимание тот факт, что по данным компьютерной томографии (КТ) органов грудной и брюшной полости и забрюшинного пространства с контрастированием было выявлено образование правой почки 19×14×24 мм с накоплением контрастного препарата. Не исключался злокачественный генез данного образования.
Характер изменений в сердце как по данным Эхо-КГ, так и по данным МРТ не исключал поражения сердца в рамках амилоидоза. Выполнена биопсия десны. Окраска на амилоид положительная.
Параллельно пациентке проводилась симптоматическая терапия (неоднократные плевральные пункции и дренирование плевральной полости) с кратковременным положительным эффектом. По данным цитологического исследования плевральной жидкости был выявлен реактивно-геморрагический выпот с выраженной лимфоидной инфильтрацией и присутствием клеток мезотелия. Микобактерий туберкулеза не найдено.
Основной диагноз: Амилоидоз с преимущественным поражением сердца. ГКМП без обструкции выносящего тракта левого желудочка.
Осложнения: хроническая сердечная недостаточность II Б, III ФК. Гидроперикард. Рецидивирующий правосторонний гидроторакс. Пункция и дренирование правой плевральной полости.
Сопутствующий диагноз: Объемное образование правой почки.
Для окончательной верификации диагноза пациентка госпитализирована в ревматологическое отделение ГКБ № 52. При поступлении состояние больной средней тяжести, кожный покров обычной окраски и влажности, повышенной плотности (не собирается в складку). Высыпаний нет. В правой половине грудной клетки на уровне пятого межреберья по заднеподмышечной линии гранулирующая рана, заживающая вторичным натяжением. Периферические лимфатические узлы не увеличены. Пастозность голеней. Температура тела в норме. В легких выслушивается везикулярное дыхание, хрипов нет, ослабление дыхания над нижними отделами справа. Частота дыхательных движений 17 в минуту. Тоны сердца приглушены, ритм правильный. Короткий систолический шум на верхушке. Частота сердечных сокращений 96 уд./мин. Артериальное давление сидя 90/60 мм рт. ст. Живот мягкий, при пальпации безболезненный. Печень не выступает из-под края реберной дуги (ординаты Курлова 7×9×10 см), селезенка не пальпируется. Стул регулярный. Мочеиспускание свободное, безболезненное. Патологии суставов не выявлено.
В лабораторных данных: общий анализ крови без патологии, в биохимическом анализе крови обращали на себя внимание гипопротеинемия 62,0–62,9–55,5 г/л, повышение уровня лактатдегидрогеназы (ЛДГ) 276 Ед/л (норма до 248), С-реактивного белка в 2 раза. Протеинурия 0,2–0,7–0,1 г/л. Ревматоидный фактор, антинейтрофильные цитоплазматические антитела, антинуклеарный фактор, антитела к н-ДНК, криоглобулин, компоненты комплемента С3, С4 в норме. Отмечалось снижение уровня иммуноглобулинов А и М. Электрофорез белковых фракций сыворотки крови: М-градиент не выявлен. Гормоны щитовидной железы в норме.
Кровь на онкомаркеры: Са 125 42,2 (норма до 35),
СА 15–3, СА 19–9, раковоэмбриональный, альфа-фетопротеин, антиген СА 72–4 — норма. Диаскин-тест отрицательный.
Пациентка госпитализирована с клинической картиной сердечной недостаточности и ранее выявленным амилоидозом по гистологии десны, что при исключении других причин хронической сердечной недостаточности делало наиболее вероятным диагнозом системный амилоидоз, однако отсутствовали клинические признаки поражения почек, периферической нервной системы, не проводилось типирование амилоидоза. В то же время имел место рецидивирующий плеврит, дебютировавший после назначения средних доз ГКС, гистологически квалифицированный как гранулематозный с клетками Лангханса. Исключались саркоидоз, туберкулез, системное заболевание соединительной ткани. Во время госпитализации течение заболевания осложнилось эмпиемой плевры, что потребовало массивной антибактериальной терапии и отсрочило морфологическую верификацию новообразования правой почки. Кроме того, по первичному заключению гистологии трепанобиопсии данных за заболевание крови не получено, однако иммунохимический анализ крови и мочи выявил моно- и олигоклональную секрецию легких цепей, что рассматривалось как косвенное подтверждение AL-амилоидоза и трактовалось гематологом как IgM MLDUS — моноклональное лимфопролиферативное заболевание неясного значения.
Трудность представляла морфологическая верификация новообразования правой почки — нефрэктомия или резекция правой почки носила крайне высокий анестезиологический риск. Биопсия под контролем КТ и ультразвукового исследования образования правой почки потребовала повторных пересмотров с последующей диагностикой доброкачественного характера опухоли.
Таким образом, диагноз множественной миеломы, ассоциированной с амилоидозом, установлен на основании гистологического и иммуногистохимического исследований, секреции белка Бенс-Джонса λ.
Учитывая возраст пациентки, наличие системного AL-амилоидоза с преимущественным поражением сердца, в качестве терапии выбора должны рассматриваться протоколы терапии множественной миеломы (курсы VCD) для больных — кандидатов на аутологичную трансплантацию гемопоэтических стволовых клеток (ауто-ТГСК) с ее последующим выполнением.
Начат курс химиотерапии. Продолжена синдромная терапия сердечной недостаточности. Пациентка консультирована в НИИ трансплантологии и включена в лист ожидания для пересадки сердца.
Только для зарегистрированных пользователей
источник
Амилоидоз – системное заболевание, в основе которого лежат обменные нарушения, приводящие к образованию и выпадению в тканях сложного белково-полисахаридного комплекса – амилоида (фибриллярного белка с β-складчатой структурой)
Классификация амилоидоза почек:
1) первичный (идиопатический) – причины достоверно неизвестны
2) вторичный – при туберкулезе легких, хронических нагноительных заболеваниях легких (бронхоэктатическая болезнь, абсцесс легкого), остеомиелит, ревматоидный артрит, язвенный колит, опухоли, подострый инфекционный эндокардит, лимфогранулематоз и т.д.
3) наследственный (периодическая болезнь, португальский вариант амилоидоза и др.)
4) старческий – при этом отложения амилоида происходит чаще в головном мозге, аорте, миокарде, поджелудочной железе.
1) AL-амилоидоз – первичный, связанный с миеломной болезнью
2) АА-амилоидоз – вторичный амилоидоз на фоне хронических воспалительных заболеваний, а также при периодической болезни
3) ATTR – наследственно-семейный амилоидоз (семейная амилоидная полинейропатия) и старческий системный амилоидоз
4) Aβ2M – амилоидоз у больных на плановом гемодиализе
5) локализованный амилоидоз – чаще у людей старческого возраста, связан с СД 2 типа, болезнью Альцгеймера и др.
Наиболее часто биохимически определяются 3 амилоида: AL – образуется плазмоцитами и содержит легкие цепи иммуноглобулина; AA – не иммуноглобулиновый белок, синтезирующийся печенью; Aβ – обнаруживается при мозговых нарушениях, связанных с болезнью Альцгеймера.
Теории патогенеза амилоидоза почек:
а) теория локального клеточного генеза – амилоид образуется в результате нарушения и извращения белково-синтетической функции ретикулоэндотелиальной системы
б) теория диспротеиноза (органопротеиноза) – в результате диспротеинемии в плазме накапливаются грубодисперсные белковые фракции и аномальные белки (парапротеины), которые проникают в ткани и образуют амилоидную субстанцию
в) мутационная теория – в результате мутации образуется особый клон клеток – амилоидобласты, которые и продуцируют амилоид
г) иммунная теория – в образовании амилоиди играет роль взаимодействие АГ с АТ при хронических воспалительных заболеваниях
Клинические проявления амилоидоза почек – 4 стадии:
а) латентная – протекает практически бессимптомно (может выявляться гепатоспленомегалия); в БАК – устойчивая диспротеинемия (повышение α2— и γ-глобулинов); в ОАК – существенное и стойкое повышение СОЭ без признаков обострения основного заболевания; в ОАМ – преходящая, нестойкая, незначительная протеинурия, иногда микрогематурия, лейкоцитурия; функция почек не страдает
б) протеинурическая – в ОАМ характерна протеинурия с колебаниями от 0,1 до 3,0 г/сут, также могут быть микрогематурия, цилиндрурия; ОАК – умеренная анемия, значительное повышение СОЭ; БАК — гипоальбуминемия, гиперглобулинемия; гипонатриемия и гипокалиемия; гиперфибриногенемия; повышение сиаловых кислот при нормальном или пониженном уровне холестерина.
в) нефротическая – проявляется нефротическим синдромом
г) азотемическая – проявляется клиникой ХПН.
Также характерны ряд внепочечных проявлений:
а) амилоидоз сердца – рестриктивная кардиомиопатия
б) поражение ЖКТ: макроглоссия (язык увеличенный, плотный при пальпации), опухолеподобная инфильтрация стенки желудка, синдром мальабсорбции при амилоидозе кишечника, гепатомегалия (печень увеличенная, плотная, с ровным, безболезненным краем) и др.
в) полисерозит (плеврит, перитонит)
г) периферическая полинейропатия, запястный туннельный синдром (сдавление срединного нерва – резкие жгучие боли в I-III пальцах кисти и лучевой стороне IV пальца, снижение чувствительности кончиков пальцев и силы мышц кисти)
д) симметричный полиартрит с утренней скованностью
е) психические нарушения в виде деменции и др.
1. Данные анамнеза: стойко и значительно повышенная СОЭ; гепатоспленомегалия; синдром мальабсорбции; протеинурия.
2. Биопсия – необходима для верификации диагноза.
3. Лабораторная диагностика – ее данные зависят от стадии процесса (см. клинические проявления)
4. Различные инструментальные исследования: Эхо-КГ, ЭКГ, УЗИ, рентгенография и др. в зависимости от пораженных органов
Принципы лечения амилоидоза:
— ограничение синтеза предшественника амилоида
— подавление синтеза амилоида и предотвращение отложения его в тканях
— лизис тканевых амилоидных структур
- лечение фонового заболевания
- симптоматическое лечение НС и/или ХПН
1. Щадящий режим, избегание физического и эмоционального переутомления
2. Диета: белок 60-70 г/сут, исключить казеин-содержащие продукты (молоко, сыр), говядину, телятину, рекомендуется баранина, крупы
3. Колхицин (1-2 мг/сут, в зависимости от переносимости) – ингибирует синтез предшественников амилоида;
4. Унитиол 5% р-р от 3-5 до 10 мл/сут в/м 30-40 дней 2-3 раза в год – тормозит агреггацию амилоидных фибрилл
5. Аминохинолиновые производные (хлорохин по 0,25-0,5 г/сут длительно) – уменьшают образование амилоида через влияние на отдельные звенья амилоидообразования
6. Диметилсульфоксид 1-5% по 30-100 мл/сут перорально
7. Сырая печень 80-120 г/сут в течение 6-12 мес – содержит комплекс антиоксидантов, улучшает общее состояние, уменьшает размеры печени, селезенки, снижает протеинурию.
Исход амилоидоза почек зависит от осложнений (интеркуррентные инфекции, кровоизлияния, тромбозы и т.д.); продолжительность жизни – 1-3 года; причины смерти – СН (после ее возникновения продолжительность жизни около 4 мес), ХПН (после ее возникновения продолжительность жизни – менее 1 года); при вторичном амилоидозе прогноз лучше, чем при первичном; у пожилых больных прогноз всегда тяжелее.
источник
ИЛ-1 интерлейкин 1
ЛС лекарственное средство
ФНО-α фактор некроза опухоли α
ХБП хроническая болезнь почек
ХПН хроническая почечная недостаточность
СD20 основной иммунофенотипический маркер В-лимфоцитов
CD34 основной иммунофенотипический маркер гемопоэтических родоначальных клеток
hsCRP «С»-реактивный белок, определенный высокочувствительным методом
NT-proBNP N-концевой пробелок мозгового натрийуретического фактора
TRAPS аутовоспалительный периодический синдром, обусловленный наследственной аномалией рецептора к фактору некроза опухоли α
1.2014 Национальные клинические рекомендации «Диагностика и лечение АА-и AL-амилоидоза» (Научное общество нефрологов России, Ассоциация нефрологов России).
Для вторичного амилоидоза характерно более раннее начало, чем при первичном (средний возраст заболевших около 40 и 65 лет, соответственно). При этом 80% больных обращается к врачу в период возникновения протеинурии и нефротического синдрома, развившихся после длительного течения хронического воспалительного заболевания – ревматоидного артрита, остеомиелита, периодической болезни и др. Основной жалобой таких больных являются отеки различной выраженности и симптомы предрасполагающего к амилоидозу заболевания.
Наиболее тяжелая и разнообразная клиническая картина отмечается у больных AL-амилоидозом, для которого характерно генерализованное поражение.
Ведущими жалобами у таких больных являются одышка, явления ортостатизма, синкопальные состояния, обусловленные сочетанием амилоидоза сердца и ортостатической гипотензии, одновременно обычно у больных наблюдают отеки, обусловленные нефротическим синдромом и, в меньшей степени, недостаточностью кровообращения.
Характерна выраженная потеря массы тела (9-18кг) вследствие нарушения трофики мышц у больных с периферической амилоидной полинейропатией. Другой причиной снижения массы тела является моторная диарея вследствие амилоидного поражения нервных сплетений кишечника или истинного синдрома нарушенного всасывания. При осмотре больных обычно выявляют увеличение печени и/или селезенки. Печень плотная, безболезненная, с ровным краем, нередко гигантская.
Поражение почек – ведущий клинический признак АА и AL амилоидоза. При АА типе амилоидоза почки бывают вовлечены в патологический процесс практически у всех больных, при AL-типе нефропатию выявляют у 80-90%. Поражение почек наблюдают и у больных многими формами семейного амилоидоза (AFib, ALys, AGel и др.).
Клинически амилоидная нефропатия манифестирует, как правило, изолированной протеинурией и характеризуется неуклонно прогрессирующим течением с последовательной сменой стадий: протеинурическая, нефротическая, ХПН. Иногда возможно развитие ХПН без предшествующего нефротического синдрома. При AL типе амилоидоза стадийность течения амилоидной нефропатии проявляется менее отчетливо.
К особенностям амилоидоза почек относят редкость гематурии и лейкоцитурии, а также артериальной гипертензии, которую даже при ХПН отмечают лишь у 20% больных АА типом амилоидоза и ещё реже при AL типе амилоидоза. Нефротический синдром и большие размеры почек сохраняются даже при развитии и прогрессировании ХПН.
Механизмы прогрессирования амилоидной нефропатии до настоящего времени полностью не изучены. Известно, что функция почек при амилоидозе коррелирует с выраженностью тубуло-интерстициального повреждения, ведущего к развитию интерстициального фиброза (выявлена зависимость между относительной площадью интерстиция, отражающей степень фиброза, и концентрацией креатинина в крови, а также обратная связь между площадью интерстиция и величиной клубочковой фильтрации).
Выраженность фиброза почечного интерстиция, в свою очередь, коррелирует с величиной протеинурии и в большей степени зависит от количества амилоида в клубочках, чем в интерстиции. Эти данные позволяют предположить общность некоторых механизмов прогрессирования амилоидной нефропатии и хронического гломерулонефрита.
Определённый вклад в прогрессирование почечной недостаточности у больных амилоидозом вносит и артериальная гипертензия, усугубляющая имеющееся повреждение клубочков из-за развития ишемических изменений.
Поражение сердца отмечают у подавляющего большинства больных AL типом амилоидоза и у части пациентов с АTTR типом амилоидоза, для АА типа амилоидоза этот симптом не характерен. В результате замещения миокарда амилоидными массами развивается рестриктивная кардиопатия.
Клинически определяют кардиомегалию, рано развивается сердечная недостаточность (у 22% больных уже в дебюте болезни), которая быстро прогрессирует и почти у 50% пациентов, наряду с аритмиями, бывает причиной смерти. Особенностью сердечной недостаточности при первичном амилоидозе служит её рефрактерность к терапии.
Нарушения ритма и проводимости при AL типе амилоидоза многообразны:
- мерцательная аритмия,
- наджелудочковая тахикардия,
- синдром преждевременного возбуждения желудочков,
- различные блокады и синдром слабости синусового узла.
Вследствие отложения амилоида в коронарных артериях возможно развитие инфаркта миокарда, обнаруживаемого на аутопсии у 6% больных. Амилоидные отложения в клапанных структурах симулируют картину клапанного порока.
Основным признаком амилоидоза сердца на ЭКГ бывает снижение вольтажа зубцов комплекса QRS. Часто выявляют инфарктоподобный тип ЭКГ.
Наиболее адекватным методом выявления признаков амилоидной кардиомиопатии считают ЭхоКГ, с помощью которой можно диагностировать симметричное утолщение стенок желудочков, дилатацию предсердий, утолщение клапанов с регургитацией крови, выпот в полости перикарда, признаки диастолической дисфункции миокарда (наиболее характерен рестриктивный тип нарушения диастолической функции – Е/А более 2).
Серьёзным патологическим признаком при AL типе амилоидоза служит ортостатическая артериальная гипотензия, которую наблюдают у 11% больных в момент установления диагноза. Обычно этот симптом связан с дисфункцией вегетативной нервной системы (амилоидоз нервных сплетений сосудов) и в тяжёлых случаях сопровождается синкопальными состояниями. Артериальная гипотензия бывает также у больных АА типом амилоидоза.
Поражение дыхательной системы чаще отмечают при AL-типе, особенно при локальном трахеобронхиальном варианте AL-амилоидоза. В большинстве случаев оно протекает бессимптомно или со скудной клинической симптоматикой. При AL-типе амилоидоза одним из ранних признаков болезни может быть охриплость или изменение тембра голоса вследствие отложения амилоида в голосовых связках, опережающего его появление в дистальных отделах дыхательных путей. В лёгких амилоид откладывается преимущественно в альвеолярных перегородках (что приводит к развитию кашля и одышки) и стенках сосудов. Описаны также ателектазы и инфильтраты, иногда опухолевидные в лёгких. Рентгенологическая картина неспецифична, смерть от прогрессирующей дыхательной недостаточности наступает редко.
Поражение органов пищеварения наблюдают при амилоидозе в 70% случаев, причём у больных AL- и АА-типами амилоидоза частота поражения тех или иных отделов ЖКТ различна.
У 25% больных первичным амилоидозом отмечают амилоидное поражение пищевода, проявляющееся преимущественно дисфагией, которая может быть одним из ранних симптомов заболевания. Поражение желудка и кишечника может проявляться изъязвлениями и перфорацией их стенок с возможным кровотечением, а также препилорической обструкцией желудка или механической кишечной непроходимостью из-за отложения амилоидных масс. У больных с преимущественным поражением толстой кишки возможно появление клинических симптомов, имитирующих язвенный колит.
Наиболее частым желудочно-кишечным проявлением AL-амилоидоза, отмечаемым почти у 25% пациентов, бывает тяжелая моторная диарея с вторичным нарушением всасывания. Возможной причиной тяжёлой диареи при этом, наряду с инфильтрацией кишечной стенки амилоидом, в том числе, и ворсин, у больных AL-типом амилоидоза служит дисфункция вегетативных нервных сплетений кишечника. Истинный синдром нарушенного всасывания развивается приблизительно у 4-5% больных. При АА-амилоидозе эти симптомы иногда также возможны, в том числе как единственное клиническое проявление амилоидоза.
Поражение печени при АА- и AL-типах амилоидоза наблюдают практически в 100% случаев, при этом обычно отмечают небольшое увеличение печени и 3-4-х кратное повышение γ-глютамилпептидазы и щелочной фосфатазы. Тяжелое поражение печени с выраженной гепатомегалией и развернутыми признаками тяжелого холестаза отмечается значительно реже (у 15-25% больных) и более характерно для AL амилоидоза. Несмотря на выраженную гепатомегалию, функция печени чаще остается сохранной.
Редким признаком амилоидоза печени является внутрипеченочная портальная гипертензия, которая может сочетаться с выраженной желтухой, холестазом печеночной недостаточностью и свидетельствует о далеко зашедшем поражении печени с риском пищеводного кровотечения, печеночной комы.
При некоторых вариантах семейного ALys-амилоидоза описаны тяжелые спонтанные внутрипеченочные кровотечения.
Увеличение селезенки, обусловленное амилоидным поражением, возникает у большинства больных и обычно сопутствует увеличению печени. Спленомегалия может сопровождаться функциональным гипоспленизмом, что приводит к тромбоцитозу, редким проявлением амилоидоза селезенки бывает ее спонтанный разрыв.
Поражение нервной системы, представленное симптомами периферической нейропатии и вегетативной дисфункции, отмечают у 17% больных AL типом амилоидоза и у пациентов с семейной амилоидной нейропатией разных типов (ATTR, AApoA1 и др.). Клиническая картина нейропатии при всех типах амилоидоза практически одинакова, поскольку обусловлена сходными процессами: в первую очередь дегенерацией миелиновой оболочки нервов, а также компрессией нервных стволов отложениями амилоида и ишемией в результате амилоидных депозитов в стенках сосудов.
В большинстве случаев возникает симметричная дистальная невропатия с неуклонным прогрессированием. В дебюте поражения нервной системы наблюдают, главным образом, сенсорные нарушения, в первую очередь болевой и температурной чувствительности, позже вибрационной и позиционной чувствительности, присоединяются двигательные нарушения. Трофические расстройства проявляются снижением массы тела. Ранними симптомами нейропатии бывают парестезии или мучительные дизестезии (онемения). Нижние конечности вовлекаются в патологический процесс чаще верхних.
Дисфункции вегетативной нервной системы часто манифестируют ортостатической артериальной гипотензией (см.выше), иногда с обморочными состояниями, диареей, нарушением функции мочевого пузыря, импотенцией у мужчин.
У 20% больных AL типом амилоидоза выявляют синдром запястного канала, обусловленного сдавлением срединного нерва в запястном канале амилоидом, откладывающемся в связках запястья. Клинически этот синдром проявляется интенсивными болями и парестезиями в I-III пальцах кисти с постепенной атрофией мышц тенара. К особенностям синдрома запястного канала при диализном амилоидозе относят его преимущественное развитие на той руке, где сформирована фистула, а также усиление болей во время процедуры гемодиализа, возможно, в результате развития феномена обкрадывания, индуцированного фистулой, что приводит к ишемии срединного нерва.
Поражение кожи наблюдают почти у 40% больных AL-амилоидозом. Для него характерно разнообразие проявлений, включая параорбитальные геморрагии (патогномоничны для AL амилоидоза), возникающие при малейшем напряжении (кашель, натуживание). Описаны также папулы, бляшки, узелки, пузырьковые высыпания. Нередко наблюдают индурацию кожи, аналогичную склеродермической.
Редким вариантом поражения кожи при AL-типе амилоидоза служат нарушения пигментации (от выраженного усиления до тотального альбинизма), алопеция, трофические нарушения.
Поражение опорно-двигательного аппарата редко (в 5-10% случаев) возникает у больных AL типом амилоидоза (исключая костные изменения при миеломной болезни). При этом характер тканевого отложения амилоида сходен при обоих этих типах амилоидоза: амилоид откладывается в костях, суставном хряще, синовии, связках и мышцах.
Амилоидные отложения в мышцах чаще наблюдают при AL-амилоидозе. Они проявляются псевдогипертрофией (гипертрофированный мышечный рельеф при значительном снижении мышечной силы) или атрофией мышц, затрудняющими движения, мышечными болями.
Макроглоссия — патогномоничный симптом AL типа амилоидоза, отмечаемый примерно у 20% пациентов, обусловлена выраженной инфильтрацией мышц амилоидом. В тяжёлых случаях макроглоссия затрудняет не только приём пищи и речь (язык может не умещаться в ротовой полости, больные часто поперхиваются, речь становится нечленораздельной), но и приводит к обструкции дыхательных путей. При АА амилоидозе она не развивается.
Среди других органных поражений при амилоидозе известны поражение щитовидной железы с развитием клинической картины гипотиреоза (при AL-типе амилоидоза), надпочечников с появлением симптомов их недостаточности (чаще при АА типе амилоидоза), экзокринных желез, приводящее к возникновению сухого синдрома, лимфаденопатия. Редким, описанным при AL и АTTR типах проявлением амилоидоза бывает поражение глаз.
Клиническая картина других типов амилоидоза варьирует в зависимости от основной локализации и распространенности амилоидных депозитов, которые иногда могут быть значительными и напоминать проявления AL-амилоидоза.
Клинические рекомендации:
При системном амилоидозе для диагностики амилоидного поражения органа нет необходимости проводить биопсию этого органа у больных с ранее верифицированным диагнозом амилоидоза по результатам биопсии другого органа. Однако точная диагностика возможна только с помощью морфологического исследования
Наиболее типичным проявлением амилоидоза почек является протеинурия более 0,5г/сут, чаще нефротического уровня. Иногда при множественной миеломе важное значение приобретает иммунохимическое электрофоретическое исследование мочи для отличия альбуминурии в рамках амилоидоза и протеинурии переполнения (наличие в моче белка Бенс-Джонса, реакция термопреципитации белка Бенс-Джонса не обладает достаточной информативностью). Для установления связи протеинурии с амилоидозом необходимо также исключить протеинурию, связанную с диабетической нефропатией и гипертонической почкой.
Характерным проявлением амилоидоза сердца является низкая амплитуда желудочковых комплексов на ЭКГ (менее 5мм в отведениях от конечностей). Патологические Q-зубцы у больных амилоидозом нередко являются псевдоинфарктными (вследствие электрически нейтральных отложений амилоида, имитирующих рубцовые изменения, обусловленные ИБС), однако при значительном амилоидозе коронарных артерий возможны и истинные инфаркты миокарда.
Наиболее четким указанием на амилоидоз сердца является утолщение межжелудочковой перегородки и/или задней стенки левого желудочка более 12мм при УЗИ, особенно когда эти изменения сочетаются с низкоамплитудной ЭКГ. Но вместе с тем важно исключить истинную гипертрофию миокарда левого желудочка у лиц с наличием потенциальных причин для ее возникновения (артериальная гипертензия и др.).
Рестриктивные нарушения диастолической функции и снижение фракции выброса не являются ранними проявлениями амилоидоза сердца, но характеризуют амилоидную кардиопатию в дальнейшем.
Применение МРТ с гадолинием с высокой вероятностью выявляет инфильтративный характер поражения миокарда и, хотя не является специфическим признаком амилоидоза сердца, может использоваться в качестве дополнительного его предиктора, в особенности у больных с моноорганным кардиальным амилоидозом. Не обладают специфичностью в отношении только амилоидоза и высокие уровни тропонинов и N-концевого пробелка мозгового натрийуретического фактора (NT-proBNP).
Утолщение свободной стенки правого желудочка у пациентов без легочной и артериальной гипертензии является указанием на высокую вероятность инфильтративного поражения миокарда правого желудочка, при моноорганном кардиальном амилоидозе таким пациентам может быть рекомендована биопсия миокарда.
Признаком амилоидоза печени является ее увеличение, с высокой специфичностью можно диагностировать амилоидоз печени у больных системным амилоидозом с гепатомегалией более 15см по данным компьютерной томографии. У больных амилоидозом обычно выявляют также холестаз (повышение щелочной фосфатазы и/или гамма-глютамилпептидазы в 1,5 раза по сравнению с нормой). Ложная диагностика амилоидоза печени возможна у больных с тяжелой застойной правожелудочковой
недостаточностью.
Диагностика периферической амилоидной полиневропатии основывается в первую очередь на клинической оценке неврологических проявлений – обычно выявляют различные
нарушения чувствительности. Из-за поражения преимущественно мелких немиелинизированных волокон электромиография и исследование скорости проведения нервного импульса обычно неинформативны для ранней диагностики амилоидной полиневропатии.
Поражение вегетативной нервной системы чаще всего проявляется ортостатической гипотензией разной степени тяжести. Однако систолическое артериальное давление менее 90 мм рт.ст. может быть обусловлено низким сердечным выбросом у больных с сердечной недостаточностью или гиповолемией у больных с тяжелым нефротическим синдромом. Другими частыми проявлениями поражения вегетативной нервной системы являются моторная диарея и дисфункции мочевого пузыря.
Диарея вследствие инфильтрации амилоидом стенки желудочно-кишечного тракта возникает редко, такую диарею трудно дифференцировать от моторной диареи в рамках
поражения вегетативной нервной системы. Наиболее надежно вовлечение желудочно-кишечного тракта при амилоидозе устанавливают по результатам морфологического исследования.
Однако обнаружение амилоида только в стенках сосудов ЖКТ еще не является критерием поражения ЖКТ, необходимо обнаружение амилоидных депозитов в интерстиции подслизистого слоя кишечника.
Нодулярный легочный и трахеобронхиальный амилоидоз за редким исключением являются проявлением локального AL-амилоидоза. Для системного AL-амилоидоза характерно
обнаружение диффузного интерстициального легочного амилоидоза. В связи с редкостью дыхательной недостаточности необходимости в морфологической верификации легочного амилоидоза обычно не возникает. Наиболее информативным методом клинического выявления амилоидоза легких служит компьютерная томография. Однако затруднения возникают при дифференцировании амилоидоза легких от поражения вследствие застойной сердечной недостаточности.
Самостоятельное значение имеет констатация амилоидоза плевры, указанием на который является рецидивирующий плевральный выпот, независимо от эффективности лечения отечного синдрома, обусловленного сердечной недостаточностью и нефротическим синдромом. При амилоидном поражении плевры жидкость, полученная во время пункции плевральной полости, нередко содержит примесь крови. При амилоидозе плевры эвакуации плеврального выпота как правило неэффективны из-за нового быстрого его накопления.
Поражение мягких тканей характерно для AL-амилоидоза. При этом амилоидная макроглоссия с инфильтрацией дна ротовой полости, периорбитальная пурпура (и кожные геморрагии на теле) патогномоничны для этого типа амилоидоза. Возможны также псевдогипертрофия скелетных мышц (с развитием мышечной слабости), лимфаденопатия, амилоидоз височной артерии.
Целесообразно перечисление в диагнозе пораженных органов, в особенности, амилоидоза сердца для оценки тяжести больных и риска быстрого прогрессирования. Признаками прогрессирования амилоидоза сердца являются дальнейшее утолщение миокарда (на 2мм и более), увеличение класса сердечной недостаточности, снижение фракции выброса на 10% и более. Наиболее информативными показателями тяжести амилоидоза сердца являются уровни NT-proBNP (в особенности более 1800нг/л), а также тропонинов (тропонин Т более 0,025нг/мл). Критериями почечного прогрессирования являются увеличение протеинурии (на 50% от исходного уровня, как правило на 1г/сут и более), уровня сывороточного креатинина (на 25% и более от исходного).
Информативным критерием прогрессирования амилоидоза печени является увеличение уровня щелочной фосфатазы на 50% от исходного. Прогрессирование полинейропатии устанавливают по результатам электромиографии и исследования скорости проведения нервного импульса (изменения возникают на продвинутых стадиях полиневропатии). Важным показателем тяжести больных AL-амилоидоза является разница в содержании свободных легких цепей иммуноглобулинов более 180мг/л, установленная методом Freelite.
- Диагностика амилоидоза с учетом клинической картины основывается на результатах морфологического исследования
- С целью выявления амилоида необходимо окрашивание препаратов ткани красителем конго красный и последующей микроскопией в поляризованном свете.
- Окончательный диагноз амилоидоза устанавливают при выявлении конгофильных масс, обладающих способностью к яблочно-зеленому или желтоватому свечению в поляризованном свете.
- Для более точной диагностики амилоидоза применяют также метод окраски тиофлафином Т, который дает светло-зеленое свечение амилоида.
- При системном амилоидозе результативна биопсия прямой или двенадцатиперстной кишки (с захватом подслизистого слоя). Наиболее эффективна биопсия пораженного органа.
Скрининг АА-амилоидоза следует проводить в следующих группах риска:
- серопозитивные и серонегативные хронические полиартриты (ревматоидный артрит, анкилозирующий спондилоартрит, ювенильный хронический артрит, псориатическая
артропатия, синдром Рейтера и др.), - хронические воспалительные заболевания кишечника (болезнь Крона, хронический язвенный колит),
- аутовоспалительные заболевания (подагра тяжелого рецидивирующего течения, семейные периодические лихорадки — периодическая болезнь, криопиринопатии, TRAPS,
гипериммуноглобулинемия D), - хронические нагноения (туберкулез, бронхоэктатическая болезнь, остеомиелит и др.),
- злокачественные солидные опухоли.
Риск АА-амилоидоза у больных хроническими воспалительными заболеваниями повышается при персистирующем повышении маркеров острой фазы воспаления («С»-реактивный белок, SAA), наличие анемии хронических заболеваний (с повышением уровня ферритина крови), особенно в сочетании с суставным синдромом (синовиты).
Для выявления олигосекреторной моноклональной гаммапатии необходимо одновременное применение метода электрофореза, метода иммунофиксации крови и суточной мочи и количественной оценки уровня свободных легких цепей иммуноглобулинов в сыворотке крови методом Freelite.
Высокая частота олигосекреторных моноклональных гаммапатий у лиц старше 50 лет требует скринингового обследования этой группы лиц на предмет моноклональных гаммапатий. Наиболее чувствительным и недорогим турбидиметрическим методом для скрининговой диагностики являет Freelite-метод количественной оценки уровня свободных легких цепей иммуноглобулинов.
Диагностика плазмаклеточной дискразии предполагает выявление моноклональной гаммапатии и оценку количества плазмацитов костного мозга, а также их структурных особенностей. Применение цитогенетического исследования и иммунофенотипирования плазмацитов важно для уточнения их клональности и злокачественности.
Все больные с плазмаклеточными дискразиями и лимфопролиферативными заболеваниями входят в группу риска AL-амилоидоза.
источник
Современные представления о периодической болезни и клинические рекомендации по диагностике и лечению
В статье представлены современные сведения о патогенезе, клинических проявлениях, диагностике и лечении периодической болезни. Обсуждаются клинические и лабораторные методы оценки активности заболевания и подходы к лечению больных, резистентных к колхицину, на основе подавления эффектов интерлейкина-1.
Периодическая болезнь (ПБ), более известная в англоязычной литературе под названием Средиземноморская лихорадка, является древнейшим заболеванием. Первые наблюдения ПБ были описаны еще на закате средневековья. Так, H. Reimann указывает, что в 1629 г. Aubrey сообщил о своем страдании, которое проявлялось ознобом, тошнотой, рвотой, болями в животе, длящимися до полусуток и возникающими сначала каждые 2 недели, позже ежемесячно, один раз в 3 месяца, один раз в полгода [1].
Как самостоятельная нозологическая единица ПБ была выделена только в середине ХХ столетия. В 1948 г. H. Reimann на основании 6 наблюдений объединил общим термином “периодическая болезнь” ряд синдромов, включающих периодическую лихорадку, доброкачественный пароксизмальный перитонит, циклическую нейтропению и перемежающуюся артралгию [2]. Основными критериями считали периодичность и доброкачественность течения. По этой причине понятие “периодическая болезнь” стало очень широким. Благодаря работам E. S):1–112. ohar и соавт. (1967) [3], а в нашей стране – О.М. Виноградовой (1964, 1973) [4], В.А. Аствацатрян и соавт. [5] клиническая картина ПБ была очерчена более отчетливо. После открытия гена MEFV, мутации которого приводят к развитию клинических проявлений ПБ, были сформулированы окончательные критерии ПБ.
Тем не менее, сохраняет актуальность выделение особого обобщающего термина для всех периодических синдромов, клинические проявления и принципы диагностики которых сходны. Учитывая их наследственный характер, широко применяется термин семейные периодические лихорадки, которые, помимо ПБ, включают в себя криопиринопатии (семейная холодовая крапивница, синдром Макла-Уэллса, NOMID-CINCA синдром), гипериммуноглобулинемию D (дефицит мевалонаткиназы), TRAPS):1–112. (синдром, обусловленный мутацией гена рецептора к фактору некроза опухоли [ФНО]-α) и некоторые другие. Все эти заболевания характеризуются беспричинно повторяющимися приступами лихорадки в сочетании с воспалением кожи, серозных оболочек и суставов [4]. В основе патогенеза ведущую роль играют генетически детерминированные нарушения врожденного иммунитета, а механизмы специфического иммунитета – гуморального, связанного с синтезом аутоантител, и Т-клеточного, не имеют значения [5].
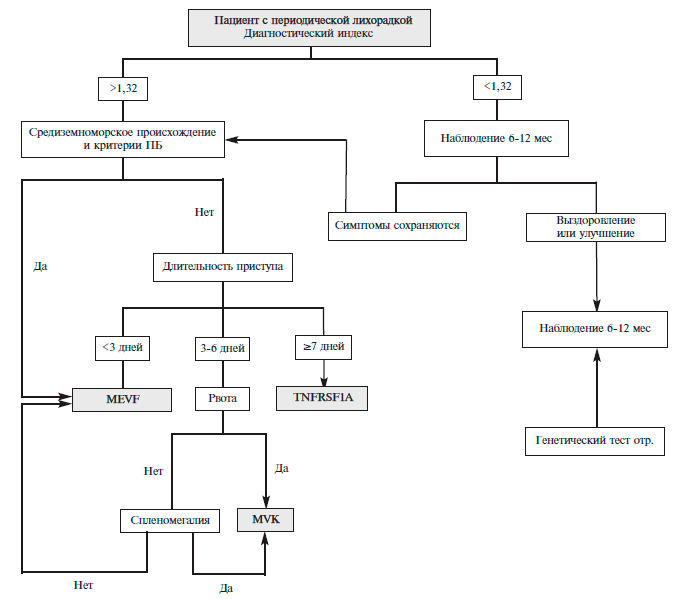 Рис. 1. Алгоритм применения диагностического индекса риска семейных периодических лихорадок
Рис. 1. Алгоритм применения диагностического индекса риска семейных периодических лихорадок
Клиническое предположение о наличии этих синдромов может возникать преимущественно у молодых пациентов с приступами болей в животе, грудной клетке в сочетании с афтозным стоматитом, диареей при наличии подобных признаков у родственников (табл. 1) [8]. Для оценки риска семейных периодических лихорадок предложен специальный индекс [9], автоматизированный расчет которого, предлагается на сайте Eurofever Project (https://www.printo.it/eurofever/index. asp). Паци ен там с высоким риском следует проводить генетическое тестирование для исключения семейных периодических лихорадок. У выходцев из Средиземно морья и Кавказа с непродолжительными приступами (менее 3 дней) в первую очередь следует исключать ПБ, у остальных пациентов с длительностью приступов до недели и эпизодами рвоты следует думать о дефиците мавалонаткиназы с развитием гипериммуноглобулинемии D, приступы длительностью более недели являются основанием для исключения TRAPS):1–112. (рис. 1). У пациентов с низким риском предлагается наблюдение за течением болезни с последующим повторным обсуждением показаний к генетическому исследованию на семейные периодические лихорадки.
| Возраст начала в мес | |
|---|---|
| Диагностический индекс = (0,067 × возраст начала) + (1,494 × абдоминалгии) – (1,504 × афтозный стоматит) + (1,958 × торакалгии) + (0,901 × диарея) + (1,503 × случаи заболевания в семье) | |
| Абдоминалгии | 0 – никогда |
| 2 – периодически или часто | |
| 3 – постоянно | |
| Афтозный стоматит | 0 – никогда |
| 1 – периодически или часто | |
| 2 – постоянно | |
| Торакалгии | 0 – отсутствуют |
| 1 – имеются | |
| Диарея | 0 – никогда |
| 1 – очень редко | |
| 2 – иногда | |
| 3 – часто | |
| Случаи заболевания в семье | 0 – отсутствуют |
| 1 — имеются | |
Одним из признаков наследственной природы ПБ является этноассоциированный характер болезни – ее широкое распространение у народов, проживающих в бассейне Средиземного моря – армян, евреев-сефардов и, реже, арабов, турков, была показана разными авторами на больших группах больных. По данным О.М. Ви но градовой лица других национальностей среди больных ПБ встречались лишь в 2% случаев [4]. Среди 86 носителей гена ПБ, наблюдавшихся J. S):1–112. amuels и соавт. [10], необычно высоким было число итальянцев и евреев-ашкенази. Однако и в этой группе 96% пациентов оказались представителями средиземноморской популяции. В группе из 150 больных ПБ, обследованных О.М. Виноградовой [4], 88,6% составили армяне. В последние годы эту болезнь на территории России широко диагностируют у азербайджанцев, представителей разных народов Северного Кавказа, имеются также отдельные наблюдения ПБ среди русских и украинцев, преимущественно жителей южных регионов России. Болезнь наследуется по аутосомно-рецессивному пути. Это означает, что у родителей симптомы заболевания обычно отсутствуют, однако в больших семьях болезнь может проявиться у родных или двоюродных братьев/ сестер, дяди или дальнего родственника.
Ген, ответственный за ПБ, был клонирован в 1997 году и обозначен аббревиатурой MEFV (MEditerranean FEver) [11,12], в том же году были идентифицированы 8 основных мутаций гена. Ген MEFV располагается на коротком плече 16 хромосомы центромерно к гену гемоглобина- α, рядом с генами, ответственными за аутосомно-доминантный поликистоз почек и туберозный склероз [12-14]. Показано, что среди евреев-сефардов, выходцев из Испании, частота носительства MEFV составляет от 1:16 до 1:8 (при распространенности ПБ в этой популяции от 1:250 до 1:1000) [15]. Частота носительства среди евреев-ашкенази Южной Европы почти на 2 порядка ниже – 1:135 (при распространенности ПБ 1:73000). Частота носительства среди американских армян составляет 1:7 [16].
Причина сохранения в современной средиземноморской популяции высокой частоты носительства MEFV остается неясной, однако сочетание ее с многочисленностью аллелей MEFV может свидетельствовать в пользу благоприятных условий для естественного отбора гетерозигот-носителей этих аллелей, по аналогии с распространенностью гена серповидноклеточной анемии в эндемичных очагах малярии.
Продуктом MEFV является белок пирин, или маренострин (от латинского Mare Nostrum – Средиземное море) [11,12]. Одним из отличительных свойств пирина является наличие В30.2-домена. Кроме того, в молекуле пирина обнаружены 2 локуса потенциального связывания с ядром, а α-спиральный и В-box-домены могут обеспечивать взаимодействие с другими белками. Все выявленные в настоящее время мутации, ассоциирующиеся с ПБ, касаются изменений в В30.2-зоне пирина.
Экспрессия MEFV происходит почти исключительно в гранулоцитах и не наблюдается в лимфоцитах и моноцитах [11]. Не обнаруживают экспрессию гена и в других тканях. Согласно основной в настоящее время рабочей гипотезе пирин является базовым регулятором воспалительного ответа нейтрофилов. Соответ ственно, структурные изменения в молекуле пирина могут изменить функцию контроля и способствовать постоянному провоспалительному потенциалу нейтрофилов.
Немутантный пирин ингибирует адапторный белок AS):1–112. C, который кроме участия в апоптозе формирует ядро инфламмасомного комплекса путем гомотипического взаимодействия с белком NLRP и каспазой, что ведет к активации интерлейкина (ИЛ)-1 β. Инфламма сома – макромолекулярная платформа в цитоплазме, устойчивая к внутриклеточным механизмам деградации белков и, в силу этого, способная обеспечить реализацию провоспалительной активности клетки. Суще ствует несколько разновидностей инфламмасом. При ПБ и других семейных периодических лихорадках основное значение придают инфламмасоме на основе белка NLRP3 – криопирина. В качестве основной причины развития ПБ рассматривают утрату ингибиторного эффекта мутантного пирина на AS):1–112. C и, как следствие, активацию каспазы-1 [17], либо формирование пирином собственной инфламмасомы [18]. Однако роль мутаций домена B30.2 в развитии ПБ остается спорной; возможно, пирин может проявлять как про-,так и противовоспалительные свойства в зависимости от конкретных условий. Значение мутантного пирина в усилении секреции ИЛ-1 β при ПБ подтверждается купированием приступа ПБ при парентеральном введении ингибиторов ИЛ-1 [19].
Таким образом, в настоящее время несомненным диагностическим критерием ПБ является выявление гомозиготного носительства мутаций пирина. Однако у 20% больных клинические признаки ПБ развиваются при наличии только одной мутации. У таких пациентов сохраняют диагностическое значение клинические критерии болезни, в частности эффективность колхицина. Окончательный диагноз в этом случае может быть установлен только через 6-12 месяцев наблюдения за эффектом колхицина.
Важное значение имеет осмотр пациента во время периодических приступов заболевания, которые проявляются лихорадкой в сочетании с абдоминалгиями, торакалгиями, артралгиями и другими, более редкими, проявлениями. Указанные проявления могут возникать в различных сочетаниях или с разной периодичностью, обычно выделяют абдоминальный, торакальный, лихорадочный, реже суставной варианты ПБ.
Болезнь возникает преимущественно в детском или юношеском возрасте, чаще у мужчин, и характеризуется хроническим течением с обострениями и ремиссиями, возникающими от разнообразных причин и через различные сроки. Между приступами болезненные проявления отсутствуют, а во время приступа болезни многие пациенты сохраняют работоспособность. Приступы продолжаются от 12 ч до 3 суток, а затем самостоятельно разрешаются. Продолжительность межприступного периода у одного и того же пациента составляет от нескольких дней до нескольких месяцев, иногда приступы следуют один за другим с промежутками в 1-3 дня, но возможны и длительные ремиссии до нескольких лет. Характерный признак болезни, имеющий диагностическое значение, – стереотипность приступов. Пациенту, как правило, без труда удается отличить боль и лихорадку, вызванные приступом болезни, от подобных симптомов другого генеза.
Лихорадка, которая у нелеченных больных, как правило, достигает высоких значений и сопровождается потрясающими ознобами, является практически постоянным симптомом приступа. Выделение особой лихорадочной формы болезни обосновывается тем, что другие симптомы, включая абдоминалгии, встречаются реже. Однако В.М. Арутюнян и соавт. полагают, что острый асептический серозит, хотя бы и кратковременный, является облигатным проявлением болезни, в связи с чем нецелесообразно расширять классификацию болезни за счет лихорадочной и суставной форм, а достаточно ограничиться тремя формами – абдоминальной, торакальной и смешанной [20].
Абдоминальный вариант наблюдается у 91% больных [11,12]. В основе абдоминального варианта ПБ лежит рецидивирующее доброкачественное воспаление серозной оболочки брюшной полости с преобладанием экссудативной реакции над пролиферативной, что подтверждается малым количеством спаек, несмотря на частоту приступов. В воспалительный процесс вовлекаются брыжейка, серозная оболочка кишки, печени, селезенки, диафрагмы. Абдоминалгии обычно сопровождаются симптомами раздражения брюшины и вялой перистальтикой, что наряду с лихорадкой и лейкоцитозом периферической крови создает существенные дифференциально-диагностические сложности и ведет к необоснованной лапаротомии.
Торакальный вариант, в основе которого лежит асептический плеврит, наблюдается у 57% пациентов [11, 12]. При рентгеноскопии грудной клетки во время приступа отмечают ограничение подвижности купола диафрагмы, небольшой выпот в синусе над диафрагмой, реже дисковидные ателектазы, все эти явления затем полностью исчезают, но у трети больных возникают спайки.
Артрит и артралгии развиваются у 45% больных [11,12], очень редко как единственное проявление болезни. Суставные проявления имеют характер летучих артралгий, моноартрита, реже полиартрита, чаще вовлекаются коленные и голеностопные суставы. По окончании приступа эти явления полностью исчезают, не оставляя пролиферативных изменений. Ревматоид ный фактор, антистрептококковые антитела не определяются.
Редко (13%) встречается рожеподобная эритема в виде болезненных плотноватых пятен диаметром 10-15 см обычно в области голеностопных суставов [11,12].
Самым тяжелым осложнением является АА-амилоидоз, строящийся из циркулирующего белка-предшественника S):1–112. AA, близкого С-реактивному белку. Частота амилоидоза значительно отличается по данным разных авторов. О.М. Виноградова на примере популяции бывшего СССР указывает частоту амилоидоза 41,3%. Аме риканские авторы, обследовавшие популяцию армян, проживающих в США, сообщают о низкой частоте амилоидоза – 2% [8,21]. В любом случае, прогноз болезни определяется наличием и тяжестью амилоидоза. Клинические проявления амилоидоза характерны для АА-типа, при котором основным органом-мишенью являются почки. В течении амилоидной нефропатии обычно удается выделить три стадии – протеинурическую, нефротическую и стадию почечной недостаточности. Особенностью амилоидоза является сохранение высокой протеинурии на стадии хронической почечной недостаточности [22]. Нелеченный ААамилоидоз в рамках ПБ обычно прогрессирует быстрее, чем при других воспалительных заболеваниях: 5- и 10летняя выживаемость составляет соответственно 48% и 24%, соответственно, а при вторичном АА-амилоидозе другой этиологии – 77% и 44% [23].
Широко распространено мнение о наследственном характере амилоидоза при ПБ. Так, у больных ПБ с отягощенным по амилоидозу семейным анамнезом риск амилоидоза увеличивается в 6 раз [24]. Описан так называемый фенотип II ПБ (некоторые исследователи ставят под сомнение наличие фенотипа II, учитывая его исключительную редкость [25]), при котором клинически выраженных приступов не бывает, однако развивается АА-амилоидоз. При этом у родственников нередко регистрируется клиника несомненной ПБ.
Благодаря обнаружению в последние годы гена ПБ и его продукта – белка пирина показаны различные механизмы наследования ПБ и АА-амилоидоза. Пирин непосредственно не участвует в метаболизме S):1–112. AA, а гены обоих белков располагаются в разных хромосомах (гены S):1–112. AA обнаружены в 11 хромосоме). Таким образом, ПБ в настоящее время, подобно другим хроническим воспалительным процессам, рассматривают в качестве пускового фактора вторичного АА-амилоидоза.
У больных ПБ описаны единичные наблюдения асептического менингита, перикардита, миалгий, узелкового полиартериита, гломерулонефрита, пурпуры Шенлейн-Геноха, клиническое значение которых не вполне ясно [4]. Спленомегалия, как правило, сопутствует амилоидозу [4].
Первая попытка создания критериев активности аутовоспалительных заболеваний была предпринята группой ученых из Клиники Шиба (Израиль) в отношении ПБ [26]. В разработанной ими шкале учитываются возраст на момент начала заболевания, частота приступов, наличие артритов, рожеподобной эритемы и доза колхицина, необходимая для достижения ремиссии болезни. В 2005 г. ученые из той же клиники провели дополнительное исследование, в котором показали недостаточность этой шкалы для корректной оценки активности болезни [27]. С использованием статистических методов были разработаны новые критерии тяжести ПБ (модифицированные критерии Тель Хашомер), которые отличаются для больных, принимающих и не принимающих колхицин [27].
Эксперты рабочих групп EUROFEVER и EUROTRAPS):1–112. попытались разработать единые критерии активности семейных периодических лихорадок с использованием дельфийского метода и метода номинальных групп [28]. Для каждого заболевания были выделены симптомы, которые, по мнению экспертов и опрошенных больных, свидетельствуют о его высокой активности. В опроснике для больных содержатся ежедневные сведения о наличии и выраженности симптомов в баллах. По завершении месяца сумма баллов делится на количество дней в месяце (30 или 31): максимальной активности соответствует 13 баллов для криопиринопатий и 16 баллов для ПБ, HIDS):1–112. и TRAPS):1–112.
В 2014 г. был завершен второй этап данного исследования [29]. Эксперты пришли к заключению, что шкала только с 2 возможными значениями для каждого признака (да/нет – 0/1 балл) проще и удобнее в применении, не отличается от изначально предложенной по чувствительности и специфичности и может быть универсальной системой оценки активности ПБ, криопиринопатий, TRAPS):1–112. и HIDS):1–112. . Оптимальный период ведения дневника, по мнению авторов исследования, составляет 3 месяца для ПБ и HIDS):1–112. , несколько меньше для криопиринопатий и больше для TRAPS):1–112. . Вне зависимости от длительности наблюдения общее число баллов делится на количество месяцев. К недостаткам предложенного метода относят необходимость длительного наблюдения, а также возможную субъективную оценку выраженности симптомов больными.
Традиционными методами подтверждения воспалительной природы заболевания и оценки его активности являются подсчет лейкоцитов крови и измерение показателей острофазового воспаления – СОЭ, уровня С-реактивного белка (СРБ), фибриногена и других.
Нередко при аутовоспалительных заболеваниях развивается анемия, как правило, нормохромная нормоцитарная, которая является следствием хронического воспаления (анемия хронических заболеваний). Веду щую роль в ее патогенезе играют провоспалительные цитокины – ФНО-α, ИЛ-1β, ИЛ-6 и интерферон-g [30,31]. Так, ФНО-α снижает эритропоэз за счет непосредственного блокирующего действия на рост эритроидных клеток-предшественниц и активации их апоптоза в костном мозге [32–34]. ИЛ-6 также подавляет костномозговую пролиферацию эритроидных клеток-предшественниц, снижает синтез эритропоэтина, через стимуляцию печеночной продукции гепсидина блокирует макрофагальное депо железа и его абсорбцию энтероцитами [35].
В последнее время исследователи стали уделять внимание новому показателю активности воспаления – отношению нейтрофилы/лимфоциты – ОНЛ (в норме оно равно 0,78-3,53 с медианой 1,65) [36]. Это надежный маркер, который может быть легко определен по результату клинического анализа крови. В настоящее время показана способность этого показателя отражать активность воспаления при ПБ. А. Ahsen и соавт. определяли ОНЛ и концентрацию СРБ у 62 больных ПБ в стадии ремиссии и 41 здорового человека [37]. Как ОНЛ, так и концентрация СРБ у больных с ремиссией ПБ были выше, чем у здоровых людей. Отмечена умеренная корреляция между этими параметрами (r=0,449, p
У больных с активным течением ПБ и других семейных периодических лихорадок отмечены более высокие уровни традиционных показателей воспаления – лейкоцитов, нейтрофилов, тромбоцитов, СОЭ, СРБ и фибриногена, по сравнению с больными, имеющими ремиссию заболевания. Однако наблюдаемые различия нередко оставались в пределах референсных значений и, следовательно, изученные показатели не могли корректно отражать активность воспаления, причем традиционные маркеры воспаления изменялись приблизительно одинаково как при аутовоспалении, так и при аутоиммунных процессах.
Реактивность S):1–112. 100A12 в сыворотке больных была существенно выше, чем стандартных маркеров воспаления. У больных ПБ с активным течением средняя концентрация S):1–112. 100A12 была почти в 3 раза выше (р=0,000059), чем у больных с ремиссией заболевания. Концентрация этого маркера в сыворотке достоверно снижалась после достижения видимого клинического эффекта терапии колхицином. Однако, даже в условиях клинической ремиссии ПБ сывороточная концентрация S):1–112. 100A12 превышала норму (120 нг/мл [51]) у всех наблюдаемых больных, что свидетельствует в пользу сохранения у этих больных остаточной активности воспаления и при отсутствии клинических проявлений. Таким образом, S):1–112. 100A12 имеет преимущество перед стандартными воспалительными маркерами по чувствительности к субклинической остаточной активности воспаления. К такому же выводу пришли А. Duzova и соавт. [44], заключив что чувствительность традиционных показателей воспаления достаточна лишь для оценки воспаления в период активности ПБ.
Выявление сохраняющейся субклинической активности воспаления при ПБ чрезвычайно важно, так как она является основной причиной развития и прогрессирования осложнений, в первую очередь, вторичного АА-амилоидоза. По-видимому, уровень S):1–112. 100A12 в крови особенно чувствителен к эффектам колхицина, так как препарат, блокируя систему микротрубочек нейтрофила, подавляет также секрецию S):1–112. 100A12, зависимую от этой системы. Патогенетический смысл этого процесса заключается также в том, что состояние системы микротрубочек определяет реализацию главного механизма повреждения ткани, связанного с выделением нейтрофилами перекисных соединений (“респираторный взрыв”). Таким образом, уровень S):1–112. 100A12 прямо отражает выраженность нейтрофильной агрессии и одновременно является индикатором полноты колхицин-зависимой блокады нейтрофила. В связи с этим по сывороточному уровню S):1–112. 100A12 можно предсказать вероятность прогрессирования амилоидоза: по данным нашего исследования у больных с прогрессирующим течением амилоидной нефропатии уровень S):1–112. 100A12 был значительно выше (р=0,039), чем у больных с медленным темпом прогрессирования.
Изменение концентрации S):1–112. 100A12 в крови позволяет не только оценить активность аутовоспаления, но и одновременно выявить роль нейтрофила в его реализации, а уровни S):1–112. 100A12 в период приступа ПБ, по-видимому, являются биохимическим эквивалентом нетоза. Высокие значения S):1–112. 100A12, характерные практически исключительно для ПБ, позволяют учитывать данный параметр при проведении дифференциальной диагностики. Концентрация S):1–112. 100A12 была ниже у больных с криопиринопатиями и TRAPS):1–112. по сравнению с больными ПБ (p=0,00014). Эти различия сохранялись и при разделении больных по активности заболеваний. Повидимому, при криопиринопатиях и TRAPS):1–112. , в отличие от ПБ, основную роль играют мак рофаги, а нейтрофил выполняет вспомогательную функцию, что проявляется менее значительным повышением концентрации S):1–112. 100A12. Тем не менее, при криопиринопатиях и TRAPS):1–112. уровень S):1–112. 100A12 также позволяет эффективно (р=0,00085) оценивать активность аутовоспаления, снижаясь в фазу ремиссии. У больных с активными аутоиммунными заболеваниями концентрация S):1–112. 100A12 также была заметно выше (p=0,0000000019), чем в ремиссию, однако не достигала значений, характерных для аутовоспалительных заболеваний. Таким образом, S):1–112. 100A12 является эффективным маркером для дифференцирования аутовоспалительных и аутоиммунных механизмов воспаления.
Несмотря на наследственный характер ПБ в настоящее время существуют эффективные методы ее лечения. С 1970 г. в практику вошел колхицин, который позволил не только предупреждать приступы болезни, но также проводить лечение и профилактику амилоидоза, что существенно улучшило прогноз больных. Механизм противовоспалительного действия колхицина может быть связан с торможением дегрануляции полиморфноядерных лейкоцитов, способностью уменьшать продукцию ИЛ-1, снижать проницаемость сосудистой стенки. Известный цитостатический эффект колхицина в применяемых дозах, по-видимому, незначителен. Ежедневный прием 1-2 мг колхицина позволяет прово дить надежную профилактику приступов ПБ и амилоидоза. Терапевтическая доза при уже развившемся амилоидозе составляет 2 мг/сут. По данным клиники им. Е.М. Тареева колхицин эффективен даже у большинства больных с нефротическим синдромом, однако эффект наступает не сразу, спустя 2-4 года постоянного приема колхицина. Эффективность препарата при амилоидозе почек резко снижается на стадии почечной недостаточности, свидетельствующей о тяжести склеротических изменений.
Указанные дозы обычно хорошо переносятся, в том числе при многолетнем приеме. Нередко развивающаяся в начале лечения диарея носит преходящий характер и, как правило, не требует полной отмены препарата.
Приблизительно у 15-20% больных колхицин оказывается изначально неэффективным. Представление о ПБ как о внешней инфламмасомопатии позволило рекомендовать назначение ингибиторов ИЛ-1 пациентам, резистентным к лечению колхицином. В Рос сийской Федерации зарегистрирован канакинумаб (Иларис®) – человеческие моноклональные IgG1 антитела к ИЛ-1, которые связываются с цитокином и блокируют его взаимодействие с рецепторами. Канакинумаб обладает длительным периодом полувыведения (21-28 дней), что позволяет вводить его подкожно каждые 8 недель. Рекомендуемая стартовая доза у взрослых составляет 150 мг один раз в 4 недели, при необходимости ее можно увеличить до 300 мг.
Эффективность и безопасность канакинумаба изучались у больных с различными аутовоспалитель ными заболеваниями. В двойном слепом, плацебоконтролируемом, рандомизированном исследовании, состоявшем из 3 час тей, 35 пациентов с криопиринассоциированным периодическим синдромом (КАПС; синдром Макла-Уэллса у 33 и синдром NOM >
Эффективность канакинумаба была подтверждена в двухлетнем открытом, многоцентровом исследовании у 166 детей и взрослых со всеми тремя фенотипами КАПС [53]. Полный ответ в течение первых 2 недель был достигнут у 85 (78%) из 109 пациентов, которые впервые начали лечение канакинумабом, в то время как у остальных пациентов отмечался частичный ответ на терапию. Во время исследования рецидивы отсутствовали у 90% больных. У этих пациентов концентрации СРБ и S):1–112. AA снизились в течение первых 8 недель после начала лечения и оставались нормальными до конца наблюдения. Эффективность препарата была в целом сопоставимой у пациентов с различными фенотипами КАПС. Следует отметить, что лечение канакинумабом ассоциировалось с нормализацией или стабилизацией аудиограмм и отсутствием прогрессирования нарушений зрения или амилоидоза почек (у 3 из 4 пациентов). Увеличение дозы канакинумаба или частоты инъекций потребовались у 24,1% больных, в основном детей и пациентов с более тяжелыми фенотипами КАПС. Основными нежелательными явлениями были инфекции, которые чаще всего были легкими или среднетяжелыми.
Эффективность канакинумаба в дозе 150 мг один раз в 4 недели (или 2 мг/кг у пациентов с массой тела ≤40 кг) в лечении ПБ, резистентной к колхицину, была установлена в двойном слепом, рандомизированном, плацебо-контролируемом исследовании CLUS):1–112. TER, в которое включали также пациентов с гипер-IgD-синдромом/дефицитом мевалонаткиназы (HIDS):1–112. /MKD) и синдромом TRAPS):1–112. [54]. В целом в этом исследовании принимал участие 181 пациент, в том числе 63 больных ПБ. Длительность двойной слепой фазы составляла 16 недель. Эффективность терапии оценивали на основании мнения врачей об активности болезни, а также лабораторных показателей (CРБ и S):1–112. AA). По частоте ответа на лечение канакинумаб достоверного превосходил плацебо. Сходные результаты были получены в двух других когортах больных, а также при анализе вторичных конечных точек (табл. 1).
| Канакинумаб | Плацебо | p |
|---|---|---|
| Частота ответа, n/N (%) | ||
| ПБ | 19/31 (61,3) | 2/32 (6,3) |