
Фенилкетонурия (фенилпировиноградная олигофрения, болезнь Феллинга) – это наследственное заболевание, связанное с нарушением обмена аминокислоты фенилаланина. В результате накопления токсических продуктов из-за неправильного метаболизма развивается отставание в умственном и физическом развитии. Причем появившиеся нарушения в состоянии здоровья необратимы, но своевременная диагностика заболевания может предотвратить все патологические изменения. Лечение заключается в исключении продуктов питания, содержащих фенилаланин. Если такая элиминационная диета применяется практически с рождения, человек вырастает здоровым. Давайте узнаем поподробнее, что же это за заболевание, чем оно проявляется, как диагностируется и лечится.
Впервые описана в 1934 г. доктором Феллингом, откуда и получила свое второе название. Встречается с частотой в среднем 1: 10 000, но в разных странах мира существуют колебания от 1:2600 (в Турции) до 1:100 000 (в Финляндии и Японии), в России от 1:5 000 до 1:10 000.

В основе заболевания лежит генетический дефект – мутация гена 12-й хромосомы (98% всех случаев фенилкетонурии). Это так называемая классическая фенилкетонурия. Ген кодирует количество фермента фенилаланин-4-гидроксилазы. Фермент отвечает за превращение аминокислоты фенилаланина в организме человека в тирозин в клетках печени. Фенилаланин – это аминокислота, которая содержится в белковых продуктах (мясо, рыба, молоко, яйца и другие).
При мутации гена количество фермента снижается, что приводит к накоплению фенилаланина и продуктов промежуточного метаболизма в тканях организма. Организм пытается избавиться от фенилаланина и продуктов его распада, выводя их с мочой.
Подобные нарушения обмена веществ приводят к нарушению строения нервных проводников, снижению образования нейромедиаторов. Все это, наряду с прямым токсическим действием избытка фенилаланина, приводит к развитию умственных нарушений, являющихся основным проявлением заболевания.
Фенилкетонурия наследуется по аутосомно-рецессивному типу, то есть не зависит от пола, и возникает при совпадении двух патологических генов от отца и матери.
Остальные 2% случаев фенилкетонурии связаны с другими генетическими дефектами и зависят от концентрации прочих ферментов (дигидроптеридинредуктазы и др.). Они имеют те же клинические проявления, но не поддаются лечению диетой. Такие варианты относят к атипичному течению заболевания. Среди них принято выделять фенилкетонурию II и III. Генетический дефект при фенилкетонурии II располагается в 4-й хромосоме, при III – в 11-й хромосоме.
Ребенок с фенилкетонурией рождается внешне здоровым, то есть ничем не отличается от других детей. С поступлением пищи в организм начинается попадание белка, а значит и фенилаланина. Последний постепенно накапливается, и обычно к 2 месяцам жизни появляются первые симптомы: вялость или беспокойство, отсутствие интереса к окружающему миру, срыгивания, изменения мышечного тонуса. Иногда срыгивания столь частые и обильные, что возникает подозрение на патологию желудочно-кишечного тракта (пилоростеноз). Ребенок из-за срыгиваний может плохо набирать в весе.
К 4-6 месяцам становится очевидной задержка психического развития. Ребенок не следит за игрушкой, не реагирует на звук, не узнает родителей. Чем дольше продолжается поступление фенилаланина в организм с едой, тем выраженнее нарушения в психической и мыслительной сферах. Развитие речи резко задерживается. Иногда словарный запас может ограничиваться несколькими словами. Если диагноз не будет выставлен и не будет начато лечение, то к 3-4 годам умственные нарушения достигнут степени идиотии (самая тяжелая степень олигофрении).
Особенностью клинического течения фенилкетонурии является необратимость возникших психических и интеллектуальных изменений. То есть при позднем выявлении помочь таким деткам уже нельзя – на всю жизнь они остаются умственно отсталыми.
Физическое развитие также отстает: дети позже начинают держать голову, переворачиваться, сидеть. Когда такие дети начинают ходить, то при этом они широко расставляют ножки, сгибая их одновременно в коленных и тазобедренных суставах. Походка покачивающаяся, мелкими шажками. В положении сидя дети принимают «позу портного» — сгибают и руки, и ноги, поджимая последние под себя. Обычно объем головы меньше, чем в норме. Может быть выраженная микроцефалия: маленькая голова.
Из других неврологических симптомов возможны нарушения мышечного тонуса, судорожные припадки. Эпилептические приступы обычно появляются в возрасте 1,5 лет и приводят к еще большему прогрессированию нарушений интеллекта.
У части больных фенилкетонурией появляются непроизвольные движения в конечностях, дрожание (гиперкинезы). В движениях нет плавности и согласованности, нарушается равновесие.
Кроме ряда психических и интеллектуальных изменений, фенилкетонурию характеризуют следующие симптомы:
- специфический «мышиный» запах (или запах плесени) от ребенка: этот симптом характерен только для фенилкетонурии. Запах появляется в результате выделения продуктов метаболизма фенилаланина (фенилпировиноградной, фенилмолочной, фенилуксусной кислот) через кожу и с мочой;
- кожные проявления: дерматиты, экзема, просто шелушение (возникают по той же причине, что и «мышиный» запах);
- позднее прорезывание зубов: у таких детей первые зубы могут появиться после 18 месяцев, эмаль недоразвита;
- нарушение пигментации: у таких детей обычно голубые глаза, очень светлая кожа и волосы в результате снижения количества меланина (его содержание зависит от метаболизма фенилаланина). Из-за этого у таких детей наблюдается повышенная чувствительность к солнечному свету;
- вегетативные симптомы: пониженное артериальное давление, повышенная потливость, запоры, акроцианоз (синюшность кистей и стоп);
- нередко фенилкетонурия сопровождается врожденными пороками сердца.
Атипичные случаи фенилкетонурии, связанные с нарушением деятельности других ферментов, участвующих в метаболизме фенилаланина, кроме умственных изменений характеризуются развитием мышечной слабости во всех конечностях с одновременным повышением мышечного тонуса, спастическим тетрапарезом. Также при этих формах развивается слюнотечение, приступы повышения температуры.
У взрослых людей, страдающих фенилкетонурией, возможно появление судорожных припадков, нарушений координации, дрожания в конечностях, ухудшения памяти и внимания, возникновение депрессии. Обычно подобные симптомы возникают при несоблюдении элиминационной диеты.
В связи с тем, что фенилкетонурия сопровождается развитием необратимых умственных нарушений, во многих странах мира, в том числе и в России, принято использовать скриниг-методы диагностики. Что это означает? Всем без исключения новорожденным детям в роддоме проводят экспресс-тесты на содержание фенилаланина. Для этого берут капиллярную кровь (из пятки) на 4-5-й день жизни ребенка (у недоношенных на 7-й), наносят на специальный бумажный бланк и отправляют в лабораторию, где по определенным изменениям врач-лаборант делает выводы о содержании фенилаланина в крови. Отрицательный тест говорит об отсутствии фенилкетонурии.
Если тест оказывается положительным, то тогда проводят дополнительные исследования для определения содержания фенилаланина в крови и моче (хроматографию, флюориметрию). Концентрацию фенилаланина в крови и моче регулярно проверяют при проведении лечения, чтобы контролировать эффективность диеты и корригировать ее при необходимости.
Возможно проведение генетического исследования для подтверждения мутации в гене, отвечающем за фенилаланин-4-гидроксилазу. Подобное исследование возможно в качестве пренатальной диагностики, то есть на этапе беременности (берут околоплодные воды путем пункции). Это инвазивное исследование делают по строгим показаниям (например, наличие больного фенилкетонурией ребенка в семье). Выявление генетического дефекта у плода позволяет прервать беременность.
На сегодняшний день самым эффективным и распространенным способом лечения фенилкетонурии является элиминационная диета: диета с исключением продуктов, содержащих фенилаланин. Если ее строго придерживаться в первые годы жизни ребенка, когда развитие нервной системы еще продолжается, то можно вырастить здорового и полноценного человека. Очень важно исключение фенилаланина именно в первый год жизни, когда наиболее активно развивается нервная система. Если элиминационная диета назначается после года, умственные нарушения не излечиваются. Каждый месяц первого года жизни без применения диеты обходится ребенку безвозвратной потерей около 4 баллов IQ. Обычно достаточно придерживаться диеты до 16-18 лет, после этого возраста организм становится менее чувствительным к токсическому действию фенилаланина, и возможно расширение рациона питания. Включение новых продуктов необходимо проводить под контролем содержания фенилаланина в крови. Иногда требуется пожизненное строгое соблюдение диеты. Беременным женщинам и женщинам, планирующим беременность, и при этом больным фенилкетонурией, для рождения здорового ребенка обязательно строгое соблюдение диеты.
Степень строгости диеты зависит от концентрации фенилаланина в крови у ребенка. При его уровне до 2-6 мг% (120-360 мкмоль/л) диета не назначается, выше этого показателя – обязательна.
Суть диеты заключается в исключении белковых продуктов.
Отказ от грудного вскармливания не обязателен, но в этом случае кормящая мать должна строго придерживаться элиминационной диеты, потому что грудное молоко содержит белок (соответственно и фенилаланин). Вопрос о возможности грудного вскармливания решается индивидуально.
 Для пополнения запасов белка назначают специальные смеси, не содержащие фенилаланин – Афенилак, Лофеналак, Нофемикс. После года это Фенилфри, Нофелан, Бигрофен, Тетрафен, МД мил ФКУ-3 и другие. В качестве прикорма назначают овощное и фруктовое пюре, фруктовые кисели, безбелковые каши (рисовая, кукурузная). После 6 месяцев можно применять специальные напитки Лопрофин, Нутриген и другие, кушать макаронные изделия, безбелковый хлеб.
Для пополнения запасов белка назначают специальные смеси, не содержащие фенилаланин – Афенилак, Лофеналак, Нофемикс. После года это Фенилфри, Нофелан, Бигрофен, Тетрафен, МД мил ФКУ-3 и другие. В качестве прикорма назначают овощное и фруктовое пюре, фруктовые кисели, безбелковые каши (рисовая, кукурузная). После 6 месяцев можно применять специальные напитки Лопрофин, Нутриген и другие, кушать макаронные изделия, безбелковый хлеб.
В России обеспечение лечебным питанием детей, больных фенилкетонурией, по закону бесплатное.
Больным фенилкетонурией противопоказаны следующие продукты: мясо, рыба (и морепродукты), орехи, творог, твердый сыр, бобовые, яйца, изделия из пшеничной муки, гречневая и манная крупа, овсяные хлопья.
Во время назначения элиминационной диеты необходим строгий контроль содержания фенилаланина в крови: первые 3 месяца жизни – каждую неделю, от 3-х месяцев до года – минимум раз в месяц, от года до 3-х лет – 1 раз в 2 месяца. Стремятся к содержанию фенилаланина 2-6 мг% у младших детей, после 10 лет – до 10 мг%. Обязательно наблюдение у детского психоневролога.
 Кроме элиминационной диеты периодически назначаются комплексы из витаминов и минералов. Если есть судорожные припадки, необходимо применение антиконвульсантов (Депакин, Клоназепам и другие). Многим из таких детей показан массаж, лечебная физкультура. Возможно использование средств физиотерапии для коррекции мышечного тонуса.
Кроме элиминационной диеты периодически назначаются комплексы из витаминов и минералов. Если есть судорожные припадки, необходимо применение антиконвульсантов (Депакин, Клоназепам и другие). Многим из таких детей показан массаж, лечебная физкультура. Возможно использование средств физиотерапии для коррекции мышечного тонуса.
Атипичные формы фенилкетонурии не поддаются лечению элиминационной диетой. В этом случае показано применение гепатопротекторов, антиконвульсантов, препаратов с Леводопой (для коррекции гиперкинезов), 5-окситриптофана, Тетрагидробиоптерина (ВН 4). Эти формы фенилкетонурии имеют худший прогноз для жизни и тем более интеллектуального развития.
На сегодняшний день разрабатываются новые направления в лечении фенилкетонурии. Среди них стоит отметить следующие:
- использование заместительной терапии фенилаланинлиазой (PAL) – растительным ферментом, расщепляющим фенилаланин до нетоксических соединений;
- генная инженерия (введение искусственно созданного нормального гена, ответственного за фенилаланин-4-гидроксилазу);
- метод «больших нейтральных аминокислот» — уменьшение всасывания фенилаланина из пищи и поступления в головной мозг с помощью специальных препаратов.
Пока эти современные разработки не имеют широкого применения, но некоторые исследования, подтверждающие их эффективность, уже проводятся.
Если женщина, страдающая фенилкетонурией, при планировании беременности и при ее наступлении не придерживается соблюдения диеты, то это отражается на развитии ее ребенка. Потомство таких женщин имеет задержку внутриутробного развития и врожденные пороки развития: пороки сердца, аномалии развития головного мозга, мочевого пузыря, микроцефалию, аномалии лицевого скелета (расщелины).
Чтобы предотвратить патологические изменения у ребенка, таким женщинам необходимо придерживаться элиминационной диеты до зачатия и всю беременность. Дефицит белка восполняется за счет специальных белковых смесей без фенилаланина.
Таким образом, фенилкетонурия – это генетическое нарушение аминокислотного обмена, которое при поздней диагностике приводит к развитию выраженных интеллектуальных нарушений у ребенка. Скрининг этого заболевания в роддомах позволяет диагностировать его в первые недели жизни и вовремя назначить лечение. Основным методом в настоящее время является назначение элиминационной диеты, которая позволяет сохранить интеллект маленькому человечку, а значит, сохранить здоровье, что обеспечит полноценное существование в течение всей жизни.
источник
2. Разберем наиболее часто встречающиеся болезни, связанные с нарушением аминокислотного обмена: фенилкетонурию и альбинизм.
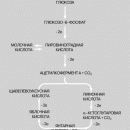
В норме аминокислота фенилаланин (ФА) с помощью фермента фенилаланингидроксилазы превращается в аминокислоту — тирозин, который в свою очередь под действием фермента тирозиназы может превращаться в меланин (пигмент). В результате нарушения активности этих ферментов развиваются два наследственных заболевания обмена человека: фенилкетонурия и альбинизм.
 фенилаланин
фенилаланин
т ирозиназа
ирозиназа


 ФЕНИЛКЕ-
ФЕНИЛКЕ-
Схема нормального обмена и его нарушений аминокислоты фенилаланина
2.1 Фенилкетонурия (ФКУ) встречается в человеческих популяциях с относительно высокой частотой — 1:10000. Заболевание наследуется по аутосомно-рецессивному типу — больные являются рецессивными гомозиготами (аа). Механизм возникновения – мутация в системе генов, отвечающих за синтез фермента фенилаланин-гидроксилазы.
Фенилаланин принадлежит к числу незаменимых аминокислот. Только часть ФА используется для синтеза белков; основное количество этой аминокислоты окисляется до тирозина. Если неактивен фермент фенилаланингидроксилаза, то ФА не превращается в тирозин, а накапливается в сыворотке крови в больших количествах и превращается в фенилпировиноградную кислоту (ФПВК), выделяющуюся с мочой и потом, вследствие чего от больных исходит «мышиный» запах. Дети с фенилкетонурией рождаются здоровыми (фенотипически), но в первые недели жизни у них появляются клинические признаки заболевания. ФПВК является нейротропным ядом, в результате чего развивается повышенная возбудимость, повышенный тонус мышц, судорожные припадки. Позже присоединяются нарушения высшей нервной деятельности, умственная отсталость, микроцефалия. У больных наблюдается слабая пигментация вследствие нарушения синтеза меланина.
Диагностика ФКУ осуществляется биохимическими методами:
еще до развития клинической картины наблюдается выделение ФПВК с мочой (хлорное железо, мазок по пеленки),
в крови определяется высокое содержание фенилаланина.
Метод лечения ФКУ является диетотерапия — снижение содержания в пище ребенка аминокислоты фенилаланина (специальные смеси с отсутствием фенилаланина, а позднее с небольшим добавление этой аминокислоты). Лечение необходимо начинать с первых недель жизни, проводить до 7—10 лет и постоянно следить за содержанием фенилаланина в крови (у человека до 7 лет идет формирование нервной системы). Мозг взрослого человека устойчив к высоким концентрациям ФПВК.
2.2. Альбинизм встречается с частотой — от 1:5000 до 1:25000. Он наследуется по аутосомно-рецессивному типу. Механизм этого заболевания — мутация гена, при которой нарушается активность фермента тирозиназы, превращающего аминокислоту тирозин в пигмент меланин.
Основными клиническими проявлениями альбинизма являются отсутствие меланина в клетках кожи (молочно-белый ее цвет), волосах (очень светлые волосы), радужной оболочке глаза и повышенная чувствительность к УФ облучению, которое вызывает воспалительные заболевания кожи. У больных на коже отсутствуют какие-либо пигментные пятна, снижена острота зрения. Фенотипические признаки выражены уже у новорожденного.
Диагностика заболевания по фенотипическим признакам.
Лечение не разработано (защита от солнечных лучей, поднятие иммунитета).
2.3. Алкаптонурия встречается довольно редко (3-5/10 6 ). Наследуется по аутосомно-рецессивному типу. Алкаптонурия является следствием мутации гена регулирующего синтез оксидазы, катализирующей превращение гомогентизиновой кислоты в малеилацетоуксусную.
В основе этой патологии лежат отложения гомогентизиновой кислоты в соединительной ткани.
клиника (фенотипические проявления: потемнение мочи на воздухе до цвета охры) проявления заболевания начинаются после 40 лет, характеризуются поражением суставов конечностей и позвоночника;
источник
Фенилкетонурия — наследственное заболевание, связанное с нарушением обмена такой аминокислоты, как фенилаланин.
Причиной развития данного заболевания является генетическая мутация, в результате которой нарушается выработка фермента фенилаланин-4-гидроксилазы, который необходим для превращения аминокислоты фенилаланин в тирозин. В результате невозможности проведения этой реакции в организме накапливается фенилаланин и продукты его метаболизма. Эти вещества в свою очередь токсичны для нервной системы. Они нарушают процессы белкового и жирового обмена, выработку гормонов, транспорт аминокислот, что особенно негативно сказывается на развитии и функционировании головного мозга.
Отмечается, что фенилкетонурия чаще встречается у девочек и у детей, где родители состоят в близкородственном браке. Тип наследования заболевания аутосомно-рецессивный.
Симптомы фенилкетонурии проявляются не сразу после рождения, а по мере накопления в организме фенилаланина и продуктов его метаболизма. Чаще всего первые проявления заболевания развиваются в возрасте 2 месяцев, при отсутствии лечения к 6 месяцам картина становится более отчетливой.
Первыми тревожными сигналами становятся вялость и апатия у малыша, хотя ребенок и наоборот может быть тревожным, легко возбудимым, возможны эпизоды рвоты. Кроме того со временем могут присоединяться судорожные припадки по типу эпилептиформных, наблюдаться гипертонус мышц, гиперкинезы, тремор конечностей, атаксия. Также специфическим признаком является своеобразный «мышиный» запах, исходящий от ребенка.
К 6-ти месяцам становится очевидным отставание в психическом развитии, вплоть до развития олигофрении и идиотии.
В физическом развитии отставание не столь явное, но тоже есть. Оно выражается уменьшением размера черепа, более поздним прорезыванием зубов, нарушением ползания и хождения. Походка у них тоже своеобразная: дети ходят мелкими шажками, раскачиваясь, а стоят с широко раздвинутыми ногами, согнутыми в тазобедренных и коленных суставах.
Поскольку развитие заболевания можно предотвратить, принципиально важна своевременная диагностика. Поэтому всем детям еще в роддоме проводится скрининговое исследование капиллярной крови. Доношенных детей обычно тестируют на 4-5 день, а недоношенных на седьмой. Маркером служит концентрация фенилаланина в крови. При изменении показателей детей направляют в медико-генетическую консультацию, а также под наблюдение педиатра и гастроэнтеролога.
Основное лечение состоит в соблюдении специальной диеты после постановки диагноза. Если заболевание выявлено не сразу, то можно только предотвратить усугубление симптомов, но полностью восстановить здоровье ребенка уже не получится.
Изначально проводится диета с исключением фенилаланина, который содержится в белковых продуктах (мясе, яйцах, субпродуктах, молочных продуктах, рыбе). Кроме того исключаются бобовые, орехи, грибы, шоколад, крупы, хлебобулочные изделия. Вместо них используются белковые гидролизаты и аминокислотные смеси. Ребенок может есть фрукты, овощные блюда. Источниками жиров являются растительное и сливочное масло. Углеводы также могут быть включены в диету. Постепенно добавляется и белковая пища в ограниченных количествах.
Для корректировки диеты и оценки состояния необходим регулярный контроль над уровнем фенилаланина в крови. Сначала исследования проводятся еженедельно, а по мере стабилизации состояния раз в месяц. В возрасте после года достаточно проведения проб 1 раз в 2-3 месяца.
Особенно строгая диета должна соблюдаться в период активного развития мозга – обычно до 12-14 лет. После рекомендации врачей разнятся: есть мнение, что больные фенилкетонурией должны пожизненно соблюдать строгую диету, другие специалисты считают, что после пубертатного возраста пациенты могут переходить на обычную пищу. Кроме того, в ряде стран ведутся исследования по разработке лекарственного препарата, применение которого поможет существенно облегчить диету.
При своевременно начатом лечении прогноз благоприятный. В нашей стране проводится поддержка семей, где есть больные фенилкетонурией. В частности, дети обеспечиваются специальным лечебным питанием вплоть до 18 лет, наблюдаются в медицинских учреждениях и при оформлении инвалидности, обеспечиваются социальными льготами.
источник
1. Альбинизм (а) и фенилкетонурия (ФКУ – заболевание, связанное с нарушением обмена веществ – в) наследуются у человека как рецессивные аутосомные признаки. Отец альбинос и болен ФКУ, а мать – дигетерозиготна по этим генам (гены, определяющие эти признаки, расположены в разных парах аутосом). Составьте схему решения задачи. Определите генотипы родителей, возможного потомства и вероятность рождения детей, не страдающих ФКУ и альбинизмом.
А – отсутствие альбинизма аВ ав
в – ФКУ F1 Cоставим решётку Пеннета
 В – отсутствие ФКУ
В – отсутствие ФКУ



 ♀
♀
 ав здоров ФКУ альбинизм альбинизм и
ав здоров ФКУ альбинизм альбинизм и
Ответ: 1) Генотипы родителей Р: мать — ♀ АаВв, отец — ♂ аавв; 2) Генотипы возможного потомства F1 АаВв – 25%, Аавв – 25%, ааВв – 25%, аавв – 25%. 3) Вероятность рождения детей, не страдающих ФКУ и альбинизмом – 25%, не страдающих альбинизмом – 50 %, не страдающих ФКУ – 50%.
2. Существуют два вида наследственной слепоты, каждый из которых определяется своим рецессивным геном (а или в). Оба аллеля находятся в различных парах гомологичных хромосом и не взаимодействуют друг с другом. Бабушки по материнской и отцовской линиям имеют различные виды слепоты. Оба дедушки хорошо видят (не имеют рецессивных генов). Определите генотипы бабушек и дедушек, генотипы и фенотипы их детей и внуков, рассчитайте вероятность рождения внуков слепыми, Объясните полученные результаты.
 Дано: Решение:
Дано: Решение:
А – нормальное зрение 1) Р ♀ААвв Х ♂ААВВ
В – нормальное зрение F1 ААВв ААВв ААВв ААВв
 в – слепота н. н. н. н.
в – слепота н. н. н. н.
2) Р ♀ ааВВ Х ♂ААВВ 3) F1 ♀ААВв Х ♂АаВВ
G аВ аВ АВ АВ G АВ Ав АВ аВ
F1 АаВВ АаВВ АаВВ АаВВ F2
 н. н. н. н.
н. н. н. н.
| ♂ ♀ | АВ | аВ |
| АВ | ААВВ Н. | АаВВ Н. |
| Ав | ААВв Н. |  АаВв Н. АаВв Н. |
Ответ: 1) 1-я семья. Родители Р: бабушка – Аавв, дедушка – ААВВ, дети (потомки первого поколения) F1: ААВв. 2) 2-я семья. Родители Р: бабушка – ааВВ, дедушка – ААВВ, дети (потомки первого поколения) F1: АаВВ. 3)
Внуки (потомки второго поколения) F2: ААВВ, ААВв, АаВВ, АаВв. Вероятность рождения слепых внуков равна 0 (слепых среди внуков не будет). Доминантный признак (нормальное зрение) будет подавлять рецессивный (слепота).
3. У кошек короткая шерсть сиамской породы доминирует над длинной шерстью персидской породы, а чёрная окраска шерсти персидской породы доминирует по отношению к палевой окраске сиамской. Скрещивались сиамские кошки с персидскими. При скрещивании гибридов между собой во втором поколении получено 48 котят. 1) Сколько типов гамет образуется у кошки сиамской породы? 2) Сколько разных генотипов получилось в F2? 3) Сколько разных фенотипов получилось в F2? 4) Сколько котят F2 похожи на сиамских кошек? 5) Сколько котят F2 похожи на персидских кошек?
 Дано: Решение:
Дано: Решение:
А – короткая шерсть Р ♀ААвв Х ♂ааВВ
а – длинная шерсть G Ав Ав аВ аВ
В – чёрная окраска F1 АаВв АаВв АаВв АаВв
в – палевая окраска к. ч. к. ч. к. ч. к. ч.
 F1 ♀АаВв Х ♂АаВв
F1 ♀АаВв Х ♂АаВв
Р — ?, F1 — ?, F2 — ? G АВ Ав АВ Ав · 48 котят аВ ав аВ ав
| ♂ ♀ | АВ | Ав | аВ | ав |
| АВ | ААВВ К. ч. | ААВв К. ч. | АаВВ К. ч. | АаВв К. ч. |
| Ав | ААВв К. ч. | ААвв К. п. | АаВв К. ч. | Аавв К. п. |
| аВ | АаВВ К. ч. | АаВв К. ч. | ааВВ д. ч. | ааВв д. ч. |
| ав | АаВв К. ч. | Аавв К. п. | ааВв д. ч. | аавв д. п. |
Ответ: 1) У кошки сиамской породы образуется один тип гамет – Ав.
3) Во втором поколении образуется 9 разных генотипов. 3) Во втором поколении образуется 4 разных фенотипа:9/16- короткошерстных чёрных, 3/16- короткошерстных палевых, 3/16- длинношерстных чёрных, 1/16- длинношерстных палевых. 4) На сиамских кошек похожи 3/16 из гибридов второго поколения- F2, т. е. 48:16·3=9. 5) На персидских кошек похожи 3/16 гибридов второго поколения, т. е. 9.
4. Нормальный рост у овса доминирует над гигантизмом, а раннеспелость над позднеспелостью. Гены обоих признаков находятся в разных парах хромосом. Какими признаками будут обладать гибриды, полученные от скрещивания гетерозиготных по обоим признакам родителей? Каков фенотип родительских особей?
А – нормальный рост Р ♀АаВв Х ♂АаВв
а – гигантизм G АВ Ав АВ Ав
В – раннеспелость аВ ав аВ ав
| ♂ ♀ | АВ | Ав | аВ | ав |
| АВ | ААВВ н. р. | ААВв н. р. | АаВВ н. р. | АаВв н. р. |
| Ав | ААВв н. р. | ААвв н. п. | АаВв н. р. | Аавв н. п. |
| аВ | АаВВ н. р. | АаВв н. р. | ааВВ г. р. | ааВв г. р. |
| ав | АаВв н. р. | Аавв н. п. | ааВв г. р. | аавв г. п. |

 F1
F1
Ответ: 1) У гибридов будет наблюдаться расщепление в отношении 9:3:3:1 – 9/16- раннеспелых с нормальным ростом, 3/16- позднеспелых с нормальным ростом, 3/16- раннеспелых гигантских, 1/16- позднеспелых гигантских. 2) Фенотип родительских особей – нормальный рост и раннеспелость.
5. У львиного зева красная окраска цветка неполно доминирует над белой. Гибридное растение имеет розовые цветы. Нормальная форма цветка полностью доминирует над пилорической. Какое потомство получится от скрещивания двух дигетерозиготных растений?
 Дано: Решение:
Дано: Решение:
| ♂ ♀ | АВ | Ав | аВ | ав |
| АВ | ААВВ К. н. | ААВв К. н. | АаВВ Р. н. | АаВв Р. н. |
| Ав | ААВв К. н. | ААвв К.п. | АаВв Р. н. | Аавв Р. п. |
| аВ | АаВВ Р. н. | АаВв Р. н. | ааВВ Б. н. | ааВв Б. н. |
| ав | АаВв Р. н. | Аавв Р. п. | ааВв Б. н. | аавв Б. п. |
 F1 — ?
F1 — ?
Ответ: От такого скрещивания получится 6 различных фенотипов: 3/16- с красными нормальными цветами, 6/16=3/8- с розовыми нормальными цветами, 3/16- с белыми нормальными цветами, 2/16=1/8- с розовыми пилорическими цветами, 1/16- с красными нормальными цветами, 1/16- с белыми пилорическими цветами.
Не нашли то, что искали? Воспользуйтесь поиском:
Лучшие изречения: При сдаче лабораторной работы, студент делает вид, что все знает; преподаватель делает вид, что верит ему. 9079 —  | 7217 —
| 7217 —  или читать все.
или читать все.
193.124.117.139 © studopedia.ru Не является автором материалов, которые размещены. Но предоставляет возможность бесплатного использования. Есть нарушение авторского права? Напишите нам | Обратная связь.
Отключите adBlock!
и обновите страницу (F5)
очень нужно
источник
Понятие о генных болезнях человека: фенилкетонурия, альбинизм, галактоземия, серповидно-клеточная анемия. Механизм развития, методы диагностики, профилактика генных болезней
Фенилкетонури́я (фенилпировиноградная олигофрения) — наследственное заболевание группы ферментопатий, связанное с нарушением метаболизма аминокислот, главным образом фенилаланина. Сопровождается накоплением фенилаланина и его токсических продуктов, что приводит к тяжёлому поражению ЦНС, проявляющемуся, в частности, в виде нарушения умственного развития.
Вследствие метаболического блока активируются побочные пути обмена фенилаланина, и в организме происходит накопление его токсичных производных — фенилпировиноградной и фениломолочной кислот, которые в норме практически не образуются. Кроме того, образуются также почти полностью отсутствующие в норме фенилэтиламин и ортофенилацетат, избыток которых вызывает нарушение метаболизма липидов в головном мозге. Предположительно, это и ведёт к прогрессирующему снижению интеллекта у таких больных вплоть до идиотии. Окончательно механизм развития нарушений функций мозга при фенилкетонурии остается неясным. Среди причин также предполагается дефицит нейромедиаторов мозга, вызванный относительным снижением количества тирозина и других «больших» аминокислот, конкурирующих с фенилаланином при переносе через гематоэнцефалический барьер, и прямое токсическое действие фенилаланина. Производится полуколичественным тестом или количественным определением фенилаланина в крови. При нелеченных случаях возможно выявление продуктов распада фенилаланина (фенилкетонов) в моче (не ранее 10-12 дня жизни ребенка). Также возможно определение активности фермента фенилаланингидроксилазы в биоптате печени и поиск мутаций в гене фенилаланингидроксилазы. Для диагностики 2 и 3 типа, связанных с мутацией в гене, отвечающем за синтез кофактора, необходимы дополнительные диагностические исследования. В возрасте от 2-4 месяцев у больных появляются такие симптомы, как вялость, судороги, экзема, мышиный запах.
При своевременной диагностике патологических изменений можно полностью избежать, если с рождения и до полового созревания ограничить поступление в организм фенилаланина с пищей.
Позднее начало лечения хотя и даёт определённый эффект, но не устраняет развившихся ранее необратимых изменений ткани мозга.
Некоторые из современных газированных напитков, жевательных резинок и лекарственных препаратов содержат фенилаланин в форме дипептида (аспартам), о чём производители обязаны предупреждать на этикетке. При рождении ребёнка в роддомах на 3-4 сутки берут анализ крови и проводят неонатальный скрининг для обнаружения врожденных заболеваний обмена веществ. На этом этапе возможно обнаружение фенилкетонурии, и, как следствие, возможно раннее начало лечения для предотвращения необратимых последствий. Лечение проводится в виде строгой диеты от обнаружения заболевания как минимум до полового созревания, многие авторы придерживаются мнения о необходимости пожизненной диеты. Диета исключает мясные, рыбные, молочные продукты и другие продукты, содержащие животный и, частично, растительный белок. Дефицит белка восполняется аминокислотными смесями без фенилаланина. Кормление грудью детей, больных фенилкетонурией, возможно и может быть успешным при соблюдении некоторых ограничений[ Альбинизм — это отсутствие пигмента в коже, волосах, тканях глаза. Различают полный альбинизм, при котором пигмент отсутствует во всем организме, и частичный альбинизм, при котором пигмент отсутствует только в отдельных органах, например в глазах. При общем альбинизме вся кожа, в том числе и кожа век, имеет бледно-розовую или молочно-белую окраску. Брови и ресницы тоже обесцвечены, белы. Изменения при альбинизме не ограничиваются кожей, а проявляются большей или меньшей недостаточностью пигмента внутри глаза: в радужке, в хориоидее, в сетчатке.
Из-за альбинизма развивается фотофобия — светобоязнь, человек не может переносить яркий свет и вынужден носить темные очки или темные контактные линзы с отверстием в центре. Данное отклонение связано с отсутствием в организме пигмента, который носит название меланин. Меланин придает окраску нашей коже, волосам, глазам. Он содержится в сосудистой оболочке глаза, благодаря чему свет попадает в глаз только через зрачок. Роль меланина в глазу очень важна. Недостаточность пигмента в сетчатке приводит к возникновению особого зрительного расстройства — никталопии. Человек, страдающий этим расстройством, плохо видит при дневном освещении и лучше — при сумеречном освещении. Существуют методы, позволяющие найти в гене мутацию, ответственную за болезнь, или идентифицировать ДНК-маркер, который генетически связан с больным геном. С помощью генетических диагностических тестов, основанных на анализе ДНК, можно выявлять генетические дефекты в организме на ранних стадиях его развития и даже еще до рождения ребенка. Нетяжелые непрогрессирующие наследственные аномалии не являются основанием для ограничения деторождения, но этот вопрос всегда лучше решать со специалистом.
Галактоземия — редкое генетическое нарушение обмена веществ, при котором изменяется нормальный процесс метаболизма углеводов (сахаров) галактозы. Галактоземию не следует путать с непереносимостью лактозы, ведь эти болезни никак не связаны. Галактоземия наследуется за аутосомно-рецессивным типом и возникает из-за дефицита активности фермента, отвечающего за усвоение организмом галактозы.
ДИАГНОСТИКА И ДИФДИАГНОСТИКА
Позитивные пробы на сахар и обнаружение галактозы в моче в первые дни жизни, а также уровень ее в крови более 0,2 г/л требуют специального обследования ребенка на галактоземию.
Существуют специальные методы определения активности ферментов, превращающихся в галактозу, которые выполняются в централизованных биохимических лабораториях.
Дифференциальный диагноз проводится обычно с сахарным диабетом.
Тяжелые формы заканчиваются летально в первые месяцы жизни, при затяжном течении на первый план могут выступать явления хронической недостаточности печени или поражения центральной нервной системы.
ЛЕЧЕНИЕ И ПРОФИЛАКТИКА
При подтверждении диагноза необходим перевод ребенка на питание с исключением, главным образом, женского молока. Для этого разработаны специальные продукты: сояваль, нутрамиген, безлактозный энпит. Рекомендуются заменные переливания крови, дробные гемотрансфузии, вливания плазмы. Из лекарственных препаратов показано назначение оротата калия, АТФ, кокарбоксилазы, комплекс витаминов.
Показана высокая эффективность раннего выявления беременных в семьях высокого риска и внутриутробной профилактики, состоящей в исключении молока из диеты беременных.
Учет семей риска позволяет рано, т. е. еще в доклинической стадии, подвергнуть специальному обследованию новорожденного и при положительных результатах перевести его на безлактозное вскармливание. Для раннего выявления предложены также специальные скрининг-программы массового обследования новорожденных.
Серповидно-клеточная анемия— это наследственная гемоглобинопатия, связанная с таким нарушением строения белка гемоглобина, при котором он приобретает особое кристаллическое строение — так называемый гемоглобин S. Эритроциты, несущие гемоглобин S вместо нормального гемоглобина А, под микроскопом имеют характерную серпообразную форму (форму серпа), за что эта форма гемоглобинопатии и получила название серповидно-клеточной анемии.
Серповидно-клеточная анемия весьма распространена в регионах мира, эндемичных по малярии, причем больные серповидно-клеточной анемией обладают повышенной (хотя и не абсолютной) врожденной устойчивостью к заражению различными штаммами малярийного плазмодия. Серповидные эритроциты этих больных также не поддаются заражению малярийным плазмодием в пробирке.
В основе диагностики серповидно-клеточной анемиилежит анализ физических свойств гемоглобина. Первым и самым старым методом такого анализа является исследование т.н. «влажного мазка». При смачивании мазка крови метабисульфитом натрия эритроциты отдают кислород и под микроскопом можно увидеть характерное изменение их формы. Для большей точности через 24 часа исследование повторяют. Другой, более распространенный метод основан на выявлении гемоглобина серповидных клеток по его сниженной растворимости в некоторых буферных растворах, что определяют по мутности раствора, содержащего такой гемоглобин. Широкое использование этого метода связано с возможностью быстрого получения результатов (уже через 10–15 минут).
К сожалению, указанные методы не позволяют отличить гетерозиготное состояние от гомозиготного. В настоящее время это можно сделать только с помощью электрофореза гемоглобина, т.е. анализа его подвижности в электрическом поле. Без такого анализа невозможны ни точная диагностика, ни надежное консультирование, но для массовых обследований он слишком дорог и занимает много времени.
Хромосомные болезни, связанные с изменением числа хромосом (синдром Патау и Эдварса, болезнь Дауна, синдром Клайнфельтера, синдром Шерешевского-Тернера и др.). Генетическая основа, основные клинические проявления и методы диагностики.
К хромосомным относятся болезни, обусловленные геномными мутациями или структурными изменениями отдельных хромосом. Хромосомные болезни возникают в результате мутаций в половых клетках одного из родителей. Из поколения в поколение передаются не более 3—5 % из них. Хромосомными нарушениями обусловлены примерно 50 % спонтанных абортов и 7 % всех мёртворождений.
Все хромосомные болезни принято делить на две группы: аномалии числа хромосом и нарушения структуры хромосом.
Дата добавления: 2018-02-28 ; просмотров: 502 ; ЗАКАЗАТЬ РАБОТУ
источник
Фенилкетонурия – это наследственное заболевание, связанное с нарушением метаболизма аминокислот, в частности, фенилаланина. Это вещество вместе с его токсическими продуктами накапливается в организме ребенка и приводит к тяжелому поражению центральной нервной системы. Больные фенилкетонурией страдают от нарушения умственного развития.
Патология чаще наблюдается у девочек, чем у мальчиков. Если родители здоровы, но являются гетерозиготными носителями мутантного гена, их дети в большинстве случаев наследуют фенилкетонурию. Также заболевание присуще малышам, родившимся в результате родственных браков. По статистике фенилкетонурия у детей встречается чаще всего в странах Европы, где на 100000 новорожденных один имеет признаки отклонения, а также в Ирландии и в России. В африканских странах болезнь практически отсутствует.
У больных фенилкетонурией наблюдается нехватка фермента фенилаланин-4-гидроксилаза, обеспечивающего превращение фенилаланина в тирозин, что, собственно, и является сущностью заболевания. Накопление вещества и его производных нарушает ЦНС, белковый обмен, метаболизм гормонов, обмен липо- и гликопротеидов. Также происходит расстройство транспорта аминокислот, сбои в обмене катехоламинов и серотонина.
Несмотря на то, что фенилкетонурия наследуется, первые признаки можно заметить не с самого рождения. Только с 3-6 месяца родители замечают у малыша вялость, повышенную раздражительность, отсутствие интереса к окружающим предметам, рвоту, беспокойство.
Симптомы фенилкетонурии во втором полугодии выражаются отставанием в психическом развитии. Около 10% малышей страдают слабой степенью олигофрении, 60% — идиотией. При фенилкетонурии у детей рост может быть как и нормальным, так и недостаточным. Зубки у таких малышей прорезываются, как правило, позже, чем у здоровых, также наблюдается уменьшение черепа. Больные фенилкетонурией начинают гораздо позже сидеть и ходить. Таких людей можно узнать визуально по своеобразной позе и походке: в положении стоя ноги широко расставлены и согнуты в коленных и тазобедренных суставах, при этом голова и плечи опущены. Ходят же больные фенилкетонурией мелкими шажками, слегка покачиваясь. Из-за повышенного мышечного тонуса они вынуждены сидеть, поджав ноги.
Чаще всего симптомом фенилкетонурии является белесость кожного покрова, который полностью лишен пигмента. Волосы у больных чаще светлые, глаза – голубые. Нередко окружающие замечают своеобразный запах от страдающих фенилкетонурией, который напоминает мышиный. Также у некоторых больных наблюдаются эпилептические припадки, которые с возрастом проходят.
К симптомам фенилкетонурии относят повышенную потливость, синюшность конечностей и дермографизм, из-за чего человек становится более подвержен негативному влиянию солнца и травм. У детей, наследовавших фенилкетонурию, часто встречается тяжелая экзема, дерматит, склонность к запорам и артериальная гипотония.
Прогноз заболевания напрямую зависит от своевременности постановки диагноза и соответствующего лечения. Несмотря на то, что симптомы патологии появляются не раньше третьего месяца жизни, наличие отклонений можно определить уже сразу после рождения. Таким образом, если малыш родился доношенным, то на 4-5-й день у него берут кровь на анализ на данное заболевание. Если же малыш родился недоношенным, тогда диагностику проводят на 1-7 день. При выявлении концентрации фенилаланина более 2,2 мг, родителей и ребенка направляют на дополнительное обследование в медико-генетический центр.
На данный момент единственным лечением фенилкетонурии является диетотерапия. С раннего возраста малыш может питаться материнским молоком, но при этом матери необходимо соблюдать рекомендации врача. Специальная диета для лечения фенилкетонурии полностью исключает пищу, содержащую белок. В рационе не должно быть мяса, орехов, рыбы, яиц, творога, бобовых, круп, хлебобулочных изделий, шоколада и других продуктов. Жизненно важные элементы больные получают из специальных смесей, которые заменяют белок. Жир человек может получать из растительного и сливочного масла. Предпочтение отдается фруктам, сокам, овощам. Страдающие фенилкетонурией должны находиться под регулярным контролем психоневролога и педиатра.
При обнаружении фенилкетонурии с первых дней жизни анализ крови проводится каждую неделю до момента нормализации показателей. После этого необходимо сдавать этот же анализ каждый месяц в течение первого года жизни ребенка. У детей постарше диагностика проводится один раз в несколько месяцев.
источник
Фенилкетонурия – тяжелое наследственное заболевание, связанное с нарушением обмена аминокислот в организме. Возникает оно в связи с неработоспособностью одного из ферментов белкового обмена. При этом происходит накопление токсических веществ в организме: в крови, моче, нервной и мышечной тканях человека.
Фенилкетонурия приводит к тяжелым поражениям нервной системы, которые влекут за собой нарушения координации работы всего организма и его развития в целом. Если не выявить заболевание в первые дни после рождения, ребенок будет отставать как в умственном, так и в физическом развитии. Без надлежащего лечения человек умирает, только достигнув зрелости, от необратимых нарушений нервной системы.
Иногда человек получает инвалидность при фенилкетонурии: при нарушениях нервной системы, когда болезнь не лечилась изначально, или тогда, когда на момент постановки диагноза человеку присущи умственные и физические отклонения.
Фенилкетонурия проявляется на первом году жизни. Основными симптомами в этом возрасте являются:
- вялость ребенка;
- отсутствие интереса к окружающему;
- иногда повышенная раздражительность;
- беспокойство;
- срыгивания;
- нарушения мышечного тонуса (чаще мышечная гипотония);
- судороги;
- признаки аллергического дерматита;
- появляется характерный «мышиный» запах мочи.
В более позднем возрасте для больных фенилкетонурией характерна задержка психоречевого развития, нередко отмечается микроцефалия. При фенилкетонурии характерны следующие фенотипические особенности: гипопигментация кожи, волос, радужной оболочки глаз. У некоторых больных одним из проявлений патологии может быть склеродермия. Эпилептические приступы встречаются почти у половины больных фенилкетонурией и в некоторых случаях могут служить первым признаком болезни.
Каким образом развивается заболевание, и что это такое? Фенилкетонурия (ФКУ) связывает несколько форм нарушения обмена фенилаланина, которые сходны по своим признакам.
- Фенилкетонурия I типа (классическая). — мутация гена фенилаланингидроксилазы, расположенного в длинном плече хромосомы 12 на участке 12q22q24.1.
- Фенилкетонурия II типа — мутация структурного гена для цитозольной дигидроптерин-редуктазы, который локализируется на коротком плече хромосомы 4 (локус 4p15.3).
- Фенилкетонурия III типа — мутация структурного гена для цитозольной 6-пирувоилтетрагидроптеринсинтазы. Ген находится на длинном плече хромосомы 11 в районе q22.3-23.3.
Все формы заболевания наследуются по аутосомно-рецессивному типу. ФКУ II и III типов – это злокачественные, или атипичные формы, которые составляют от 1 до 3% от всех случаев фенилкетонурии. Патогенез предопределен неправильным обменом фенилаланина, вследствие чего наблюдается накопление в организме его токсичных производных.
Известно, что фенилпировиноградная, α-толуиловая, фенилмолочная кислоты, фенилэтиламин и ортофенилацетат, которые в норме почти никогда не образуются, появляются в биологических жидкостях организма и оказывают токсическое действие на ЦНС. Предполагается, что данное обстоятельство и ведет к снижению интеллекта, но многое в механизмах развития дисфункций мозга до сих пор полностью не выяснено.
Поражение нервной системы может быть результатом ряда факторов:
- дефицит нейромедиаторов мозга (катехоламинов и серотонина);
- прямое токсическое действие фенилаланина на ЦНС;
- нарушение в обмене белков;
- расстройство мембранного перемещения аминокислот;
- нарушение метаболизма гормонов.
Диагностировать фенилкетонурию необходимо у врача-педиатра сразу после рождения ребенка, чтобы предотвратить тяжелые последствия заболевания. Именно поэтому на 4-й -5-й день жизни доношенного новорожденного и на 7-й день – у недоношенного малыша в роддоме берется кровь для обследования на это заболевание.
После кормления через час специальный бумажный бланк пропитывается капиллярной кровью. В том случае, если в образце крови концентрация фенилаланина превышает 2,2мг%, такого ребенка направляют для дообследования, осмотра и уточнения диагноза в медико-генетический центр.
В том случае, если новорожденному по какой-то причине в роддоме не проводился анализ на фенилкетонурию, родители должны сделать это самостоятельно, так как данное заболевание успешно лечиться, если оно своевременно обнаруженно. Есть такие способы диагностировать фенилкетонурию: обнаружить в моче малыша фенилкетоны еще в роддоме или же определить их в крови и моче взрослых людей при соответственной клинической симптоматике.
Так как данная патология имеет аутосомно-рецессивный тип наследования, существует возможность выявить носителей мутантного гена и просчитать возможность рождения ребенка с фенилкетонурией. Для этого проводится тест на толерантность к фенилаланину: человек выпивает 10 гр. раствора фенилаланина, а через некоторое время определяют количество образовавшегося в крови тирозина. Если показатели становят 9-10 грамм, такой человек носителем фенилкетонурии не является.
Главным способом лечения фенилкетонурии является диетотерапия, ограничивающая поступление в организм белка и фенилаланина. Основным критерием адекватности диеты при фенилкетонурии служит уровень фенилаланина в крови, который должен:
- в раннем возрасте составлять 120–240 мкмоль/л;
- у детей дошкольного возраста – не превышать 360 мкмоль/л;
- у школьников – не превышать 480 мкмоль/л;
- у детей старшего школьного возраста допустимо увеличение содержания фенилаланина в крови до 600 мкмоль/л.
Пищевой рацион строится путем резкого ограничения поступления белковых продуктов животного и растительного происхождения и, следовательно, фенилаланина. Для облегчения расчетов принято считать, что 1 г условного белка содержит 50 мг фенилаланина.
При лечении фенилкетонурии полностью исключают продукты, богатые белком и фенилаланином: мясо, рыбу, сыр, творог, яйца, бобовые и др. В пищевой рацион больных входят овощи, фрукты, соки, а также специальные малобелковые продукты – амилофены.
Для коррекции белкового питания и восполнения недостатка аминокислот при фенилкетонурии назначаются специальные лечебные продукты:
- белковые гидролизаты: нофелан (Польша), апонти (США), лофенолак (США);
- смеси L-аминокислот, лишенные фенилаланина, но содержащие все другие незаменимые аминокислоты: фенил-фри (США), тетрафен (Россия), П-АМ универсальный (Великобритания).
Несмотря на обогащение аминокислотных смесей и белковых гидролизатов минеральными и другими веществами, больные фенилкетонурией нуждаются в дополнительном назначении витаминов, в частности группы В, минеральных соединений, особенно содержащих кальций и фосфор, препаратов железа и микроэлементов.
В последние годы для страдающих фенилкетонурией была обоснована необходимость применения препаратов карнитина (L-карнитин, элькар в средней суточной дозе 10–20 мг/кг массы в течение 1–2 мес. 3–4 курса в год) для профилактики его недостаточности.
Параллельно лечение фенилкетонурии осуществляется медикаментозным патогенетическим и симптоматическим лечением ноотропными средствами, препаратами, улучшающими сосудистую микроциркуляцию, по показаниям – антиконвульсантами.
Широко используется лечебная гимнастика, общий массаж и др. Комплексная реабилитация детей с фенилкетонурией предусматривает специальные методы педагогических воздействий в процессе подготовки к школе и школьного обучения. Больные нуждаются в помощи логопеда, педагога, в ряде случаев – дефектолога.
Большие споры вызывает вопрос о длительности диетотерапии в лечении фенилкетонурии. В последнее время большинство врачей принимает точку зрения о необходимости продолжительного выполнения диетических рекомендаций. Обследование детей, прекративших соблюдать диету в школьном возрасте, и детей, продолжавших получать диетотерапию, однозначно показало значительно более высокий уровень интеллектуального развития последних.
У больных фенилкетонурией старшего возраста, в том числе подростков, безусловно, возможно постепенное расширение диеты в связи с улучшением толерантности к фенилаланину. Коррекция питания осуществляется, как правило, путем введения в рацион ограниченного количества круп, молока и некоторых других натуральных продуктов, содержащих относительно умеренное количество фенилаланина. В период расширения рациона проводятся оценка нервно-психического статуса детей, контроль электроэнцефалограммы, уровня фенилаланина в крови.
В возрасте старше 18–20 лет проводится дальнейшее расширение диеты, однако и во взрослом периоде пациентам рекомендуется отказаться от высокобелковых продуктов животного происхождения. Особенно строго подходят к диетотерапии девочек, страдающих фенилкетонурией, и женщин в репродуктивном периоде. Такого рода больным фенилкетонурией необходимо продолжать диетическое лечение для обеспечения рождения здорового потомства.
В последние годы разрабатывается способ снижения уровня фенилаланина в крови путем приема препарата, содержащего фенилаланингидроксилазу растительного происхождения.
Народная медицина предлагает придерживаться безфенилаланиновой диеты всю жизнь. Ребенок вырастает, уже зная о том, что у него фенилкетонурия: прогноз для детей, которые придерживаются диеты всю жизнь более оптимистичный, чем для тех, кто обходится без фенилаланина только до 12 лет. Именно поэтому лучше всего следовать советам народной медицины и соблюдать правильную диету в течение всей жизни. Это шанс для больного человека прожить долгую и полноценную жизнь.
Самый главный рецепт народной медицины при диагнозе фенилкетонурия – питание должно быть насыщенно растительными белками. В растительной пище меньше фенилаланина и меньше белковая ценность. При этом фрукты и овощи содержат большое количество витаминов и микроэлементов, жизненно необходимых человеческому организму. По сути, народная медицина предлагает человеку стать вегетарианцем.
Растительная пища богата не только витаминами и микроэлементами. Белковый состав в ней меньше, поэтому для усвоения белка в достаточном количестве ее потребуется больше, чем для насыщения организма мясом или молоком. Для насыщения организма и утоления чувства голода лучше всего с растительной пищей употреблять каши и хлебобулочные изделия из муки с минимальным содержанием фенилаланина.
Фенилаланин содержится во всех продуктах, содержащих белок, поэтому пища с высоким содержанием белка должна быть исключена из рациона питания больных фенилкетонурией. В первую очередь исключить нужно молочные и мясные продукты. Исключение составляют младенцы, которые должны получать небольшую дозу материнского молока (количество молока согласовывается с врачом) вместе с искусственным детским питанием, которое не содержит фенилаланина.
источник
ОБМЕНА ВЕЩЕСТВ НАРУШЕНИЯ, возможные при любом заболевании нарушения каких-либо из многочисленных химических процессов, участвующих в обмене веществ в организме. Разнообразные патологические проявления включают изменения скорости роста, теплопродукции, выработки энергии для мышечной деятельности и энергетического обеспечения жизненно важных функций организма. Известно, однако, большое число т.н. болезней обмена веществ, или болезней метаболизма, причиной которых служит специфическое его нарушение; ниже описаны лишь наиболее важные из них.
Это врожденное нарушение обмена веществ, характеризующееся накоплением избыточного количества гликогена в тканях организма. Оно связано с недостаточностью фермента глюкозо-6-фосфатазы, который необходим для распада гликогена, в силу чего тот накапливается в тканях. Болезнь обычно проявляется уже в младенчестве отставанием в росте, выпячиванием живота из-за увеличения размеров печени и снижением уровня сахара в крови. Единственное средство лечения – диета; рекомендуется частое кормление и добавление в рацион глюкозы. С возрастом состояние ребенка постепенно улучшается.
– наследственная задержка психического развития, обусловленная недостаточностью единственного фермента – фенилаланингидроксилазы, необходимой для превращения аминокислоты фенилаланина в другую аминокислоту – тирозин. Накапливающийся фенилаланин оказывает токсическое действие на ткань головного мозга. Болезнь впервые была описана в 1934 А.Фёллингом. Она встречается с частотой 1 на 20 000 новорожденных независимо от пола и наиболее распространена среди европейцев.
Новорожденные внешне выглядят здоровыми, но в возрасте трех-четырех месяцев у них начинает проявляться отставание в психическом развитии. К 2–3 годам дети прекрасно развиваются физически, но психически отстают. Поскольку нарушение психического развития поддается лечению, крайне важна ранняя диагностика; в отсутствие лечения коэффициент интеллектуального развития (IQ) за каждые 10 недель снижается на 5 пунктов. Фенилкетонурию можно обнаружить уже в первый день жизни по результатам анализа крови или мочи новорожденного.
Единственным способом лечения является диета. Поскольку все обычные белковые продукты содержат фенилаланин (в количестве 4–6%), необходимо использовать лишенные этой аминокислоты синтетические продукты.
При нормальном метаболизме фенилаланина и тирозина (обе аминокислоты связаны между собой в обмене) образуется кожный черный пигмент меланин. Врожденное отсутствие этого пигмента в глазах, коже и волосах у лиц с альбинизмом обусловлено недостаточностью одного из ферментов метаболизма фенилаланина и тирозина.
Это заболевание вызывается генетически обусловленной недостаточностью фермента, участвующего в метаболизме гомогентизиновой кислоты – промежуточного продукта обмена фенилаланина и тирозина. Накапливающаяся гомогентизиновая кислота выделяется с мочой, придавая ей черный или коричневый цвет. В более позднем возрасте в соединительной ткани и хрящах откладывается синевато-черный пигмент и развивается артрит. В качестве лечения назначают диету, исключающую потребление фенилаланина и тирозина.
Неспособность организма разрушать холестерин и липопротеины низкой плотности (в составе которых он в основном находится) приводит к накоплению холестерина в тканях из-за чрезмерно высокого его уровня в крови. Состояние, при котором холестерин откладывается в подкожных тканях, называют ксантоматозом. Отложения холестерина в стенках кровеносных сосудов вызывают атеросклероз. При гиперхолестеринемии возможно также увеличение селезенки, печени или лимфатических узлов. Для лечения и профилактики используют диету.
Подагра и подагрический артрит – хронические заболевания, вызываемые нарушением обмена эндогенной (образующейся в организме) мочевой кислоты; ее соли (ураты) откладываются главным образом в хрящах, особенно суставных, и в почках, обусловливая болезненные воспалительные отеки. Накопление уратов можно предотвратить с помощью диеты. Для ослабления болей применяют специальные средства.
Многие процессы метаболизма непосредственно контролируются гормонами. Поэтому дисфункция эндокринных желез также может приводить к нарушениям обмена веществ. См. также ГОРМОНЫ; ДИАБЕТ САХАРНЫЙ; ЗОБ; НАСЛЕДСТВЕННОСТЬ; ПОДАГРА; ФЕНИЛКЕТОНУРИЯ.
источник