Фенилкетонурия, фенилпировиноградная олигофрения, наследственное заболевание из группы ферментопатий, в основе которого лежит аномалия аминокислотного обмена вследствие отсутствия или резкого снижения активности фермента фенилаланингидроксилазы. Описана в 1934 норв. учёным А. Фёллингом (A. Foiling) (болезнь Фёллинга). Частота фенилкетонурии – 1 случай на 10–15 тыс. новорождённых; наследуется по аутосомно-рецессивному типу. При фенилкетонурии фенилаланингидроксилаза сохраняет только около 5% активности, в связи с чем нарушается обмен фенилаланина и вследствие этого – тирозина, триптофана и др., накапливаются промежуточные продукты обмена – фенилэтиламин, фенилпировиноградная кислота и др. и возникает дефицит метаболитов, необходимых для нормального функционирования организма. В частности, вероятная причина умственных расстройств – дефицит медиаторов нервной системы (адреналина, норадреналина, серотонина и др.). Таким образом, при фенилкетонурии возникает комплекс взаимосвязанных метаболических расстройств, состоящий из первичного ферментного нарушения и обусловленных им др. нарушений обмена.
Фенилкетонурия проявляется главным образом выраженной олигофренией (идиотией или имбецильностью). Диагностируется в первые дни жизни ребёнка с помощью экспресс-методов – микробиологических или биохимических. Последние основаны на определении пировиноградной кислоты в моче посредством индикаторов (проба Фёллинга). Лечение сводится главным образом к специальной диете (резкое ограничение продуктов, содержащих фенилаланин).
Альбинизм (albinismus) представляет собой врожденное отсутствие кожного пигмента. Этиология и патогенез изучены недостаточно. Известно, что в результате нарушения синтеза ферментов тирозиназы, дофаоксидазы прекращается образование меланина из тирозина, диоксифенилаланина. О сложности механизмов происхождения альбинизма свидетельствуют нередко сопутствующие ему такие аномалии, как врожденная глухота, дефекты интеллекта, патология глаз и другие.
Выделяют тотальный, неполный и частичный альбинизм.
Тотальный альбинизм наследуется аутосомно-рецессивно со средней частотой 1: 10000-20000. Предполагают, что носители мутантного гена составляют 1,5% среди всех нормально пигментированных людей.
Депигментация кожи и придатков наблюдается с рождения, сопровождается сухостью кожи, нарушением потоотделения, иногда гипо- или гипертрихозом, особенно на открытых участках. У больных легко возникают солнечные ожоги, актинический хейлит. Они предрасположены к развитию кератом, эпителиом, телеангиэктазий. Из-за отсутствия пигмента в тканях глаза зрачки кажутся красными. Характерными являются горизонтальный нистагм и выраженная светобоязнь. Часто наблюдаются сходящееся косоглазие, снижение остроты зрения в результате нарушений рефракции, катаракты, возможна микрофтальмия. Нередко наблюдаются бесплодие, иммунодефицит (отсюда частые инфекции), пороки развития, сокращение продолжительности жизни, олигофрения.
Неполный альбинизм (син.: альбиноидизм), в отличие от предыдущей формы, наследуется аутосомно-доминантно, в некоторых случаях — рецессивно. Имеет место снижение активности тирозиназы, но не блокада ее синтеза. Наблюдается гипопигментация кожи, волос, радужки, иногда фотофобия. Других дефектов и аномалий не регистрируется.
Частичный альбинизм (син.: пиебалдизм) наследуется аутосомно-доминантно. Проявления выявляются при рождении. Характеризуется появлением участков ахромии на коже живота, лица, нижних конечностей, прядей седых волос. Депигментированные пятна неправильной формы с резкими границами, на их поверхности имеются мелкие темно-коричневые пятнышки. Вокруг ахромичных пятен кожа может быть пигментированной. Поражений других органов обычно не бывает. Частичный альбинизм является одним из проявлений синдромов Чедиака-Хигаси, Клейна-Ваарденбурга, Титце, Менде, Хермански-Пудлака, Кросса-МакКьюзика-Брина.
Дифференциальный диагноз альбинизма проводят с витилиго, синдромами Клейна-Ваарденбурга, Алеззандрини, Фогта-Коянаги-Харады.
Алкаптонурия (alcaptomiria; алкаптон (гомогентизиновая кислота) + греч. uron моч ) — наследственная болезнь, обусловленная нарушением обмена тирозина вследствие пониженной активности фермента гомогентизиназы и накоплением в тканях организма гомотентизиновой кислоты; проявляется у взрослых пигментацией различных тканей, развитием артрозов, а у детей — лишь темным окрашиванием мочи и иногда ушной серы; наследуется по аутосомно-рецессивному типу.
источник
Тирозин, помимо участия в синтезе белков, является предшественником гормонов надпочечников адреналина, норадреналина, медиатора дофамина, гормонов щитовидной железы тироксина и трийодтиронина, пигментов. Нарушения катаболизма тирозина многочисленны и называются тирозинемии.
Тирозинемия типа I (гепаторенальная тирозинемия) возникает при недостаточности фумарилацетоацетат-гидролазы . При этом накапливается фумарилацетоацетат и его метаболиты (сукцинилацетон), поражающие печень и почки. Частота заболевания 1:100-120 тыс новорожденных.
Существует две формы – острая и хроническая.
Острая форма составляет большинство случаев этой тирозинемии с началом в возрасте 2-7 мес и смертью 90% больных в возрасте 1-2 года из-за недостаточности печени.
К симптомам относится гипотрофия, рвота, «капустный запах» от тела и мочи, задержка развития, кровоточивость, диарея, мелена, гематурия, желтуха, анемия, периферические невропатии и параличи, кардиомиопатия, слабость мышц, дыхательные нарушения. Отмечают гипогликемию вследствие гиперплазии островковых клеток поджелудочной железы.
При хронической форме болезнь развивается позднее, медленнее прогрессирует. Продолжительность жизни около 10 лет.
Наблюдаются гипотрофия, узелковый цирроз печени и печеночная недостаточность, множественные дефекты почечной канальцевой реабсорбции с появлением синдрома Фанкони (щелочная рН мочи, глюкозурия, протеинурия), аминоацидурия, лейкопения, тромбоцитопения.
Из-за поражения печени и почек возникают проявления рахитоподобных заболеваний (остеопороз, остеомаляция). В результате печеночной недостаточности возникают симптомы, напоминающие острую порфирию. Непостоянными признаками являются умственная отсталость и неврологические изменения.
Лекарственным средством является нитизинон , конкурентный ингибитор 4-гидроксифенилпируват-диоксигеназы. В результате прекращается образование гомогентизиновой кислоты и ее дальнейший распад. Также используется диета со снижением количества фенилаланина и тирозина, инъекции глутатиона.
Гораздо более редкое заболевание по сравнению с тирозинемией I типа.
Тирозинемия типа II (глазокожная тирозинемия) возникает при недостаточности тирозин-аминотрансферазы .
Наблюдается задержка умственного и физического развития, микроцефалия, катаракты и кератоз роговицы (псевдогерпетический кератит), гиперкератоз кожи, членовредительство, нарушение тонкой координации движений.
Поражения почек и печени не наблюдается.
Эффективна диета с низким содержанием тирозина, при этом поражения кожи и роговицы быстро исчезают.
Тирозинемия III типа – результат генетического дефекта 4-гидроксифенилпируват-диоксигеназы . Зафиксировано лишь несколько случаев этой болезни.
Характерные особенности включают умеренную умственную отсталость, судороги и периодическую потерю равновесия и координации (прерывистая атаксия).
Тирозинемия новорожденных – результат кратковременного снижения активности 4- гидроксифенилпируват-диоксигеназы . Чаще наблюдается у недоношенных детей.
Наблюдается сниженная активность и летаргия. Аномалия считается безвредной. Дефицит аскорбиновой кислоты усиливает клиническую картину.
Диета со снижением количества белка, фенилаланина, тирозина и высокие дозы аскорбиновой кислоты (100 мг/день).
Генетическая аутосомно-рецессивная энзимопатия. В основе заболевания лежит снижение активности печеночного фермента гомогентизат-оксидазы , в результате в организме накапливается гомогентизиновая кислота.
Так как гомогентизат на воздухе окисляется и полимеризуется в меланиноподобное соединение, то наиболее частым и постоянным симптомом является темная моча, на пеленке и нижнем белье остаются темно-коричневые пятна. Другим образом в детском возрасте болезнь не проявляется.
С возрастом гомогентизиновая кислота, накапливается в соединительно-тканных образованиях, склерах и коже, вызывает шиферно-глубокий оттенок ушного и носового хрящей (охроноз), окрашивает одежду, контактирующую с потеющими участками тела (подмышки).
Из-за связывания гомогентизата с коллагеном ухудшается состояние соединительной ткани, что делает хрупкими хрящевые образования. После 30 лет развивается дегенеративный артрит позвоночника и крупных суставов (бедренные, коленные), межпозвонковые пространства сужены, снижается минеральная плотность костей. Может наблюдаться поражение почек и сердца.
Хотя эффективные способы неизвестны, по аналогии с другими аминокислотными нарушениями рекомендуется с раннего возраста ограничить потребление фенилаланина и тирозина, что должно препятствовать развитию охроноза и суставных нарушений. Назначают большие дозы аскорбиновой кислоты для снижения связывания гомогентизиновой кислоты в соединительной ткани. Предлагается использовать препарат нитизинон , конкурентный ингибитор 4-гидроксифенилпируват-диоксигеназы.
Заболевание обусловлено полным или частичным дефектом синтеза фермента тирозиназы (частота 1:20000), необходимой для синтеза диоксифенилаланина и далее меланинов в пигментных клетках.
При полном отсутствии фермента наблюдается тотальная депигментация кожи, волос, глаз, причем окраска одинакова для всех расовых групп и не меняется с возрастом. Кожа не загорает, совершенно отсутствуют невусы, какие-либо пигментные пятна, развиваются фотодерматиты. Сильно выражены нистагм, светобоязнь, дневная слепота (т.к. имеется депигментация сетчатки и ускоренный распад родопсина), красный зрачковый рефлекс.
При частичной недостаточности фермента отмечаются светло-желтые волосы, слабо пигментированные родинки, очень светлая кожа.
Рекомендуется использовать различные средства защиты от ультрафиолетовых лучей.
Биохимической причиной паркинсонизма (частота после 60 лет 1:200) является низкая активность тирозин-гидроксилазы или ДОФА-декарбоксилазы в нервной ткани, при этом развивается дефицит нейромедиатора дофамина и накопление тирамина.
Наиболее распространенными симптомами являются ригидность мышц, скованность движений, тремор и самопроизвольные движения.
Требуется систематическое введение лекарственных аналогов дофамина и применение ингибиторов моноаминоксидазы.
источник
1. Этиология, патогенез и последствия нарушений сосудистой проницаемости и транскапиллярного обмена.
Нарушения проницаемости сосудов (транскапиллярного обмена) возникают вследствие патологии самой сосудистой стенки (главным образом, эндотелия и базальной мембраны капилляров и венул), нарушения способности пропускать воду и содержащиеся в ней вещества благодаря процессам ультрафильтрации, диффузии, пиноцитоза, активности внутриклеточных переносчиков как без затраты энергии, так и с затратой. Причинами повышения проницаемости микрососудов (транскапиллярного обмена) чаще всего становятся воспалительные процессы в тканях, аллергические реакции, шок, гипоксия тканей, ожоги, сердечная недостаточность, тромбоз и сдавление вен, гипопротеинемия, трансфузия белковых и солевых растворов.
Факторами, приводящими к повреждению стенки сосуда в тканях в очаге воспаления, бывают токсины, кинины, гистамин. Они деформируют эндотелий, базальную мембрану, увеличивают межэндотелиальное пространство. Аллергические реакции и гипоксия так же сопровождаются ультраструктурными изменениями эндотелия.
В результате микротравм стенок сосудов происходит развитие ацидоза и активация гидролаз (приводящие, соответственно, к неферментному и ферментному гидролизу основного вещества базальной мембраны сосудов), набухание (отёчность) эндотелиальных клеток, появление и увеличение шероховатости (бахромчатости) их оболочек, (приводящие к расширению межэндотелиальных щелей, отделению эндотелиоцитов друг от друга и выпячиванию их в просвет сосуда), перерастяжение стенок микрососудов (приводящее к растяжению фенестр и образованию микроразрывов в стенках микрососудов).
Повреждение сосудистой стенки приводит к нарушению, как правило, увеличению транскапиллярного обмена за счёт возрастания:
• пассивного транспорта веществ через поры (каналы) эндотелиальных клеток и межэндотелиальные щели посредством возрастания простой, облегчённой и ионообменной диффузии и фильтрации (в силу увеличения концентрационного, электрохимического и гидродинамического градиентов);
• активного транспорта веществ через эндотелиальную клетку (против электрохимического и концентрационного градиентов), осуществляемого за счёт энергии метаболических процессов (т.е. с затратой энергии макроэргов.); активный транспорт веществ может осуществляться при помощи внутриклеточных переносчиков, пиноцитоза, фагоцитоза а также комбинированным путём в результате образования различных ФАВ.
При увеличении фильтрации (вследствие резко повышенной проницаемости стенок артериальной части капилляров) и ослаблении реабсорбции (в результате возрастания как гидростатического давления в венулярной части капилляра, так и коллоидно-осмотического давления межклеточных пространств) и затруднении лимфооттока наблюдают максимальный отёк межклеточных структур, сдавливающий стенки капилляров, сужающий их просвет и резко затрудняющий в них кровоток, вплоть до развития стаза.
2. Наследственные нарушения обмена аминокислот: фенилкетонурия, алкаптонурия, альбинизм. Этиология и патогенез.
Фенилкетонури́я (фенилпировиноградная олигофрения) — наследственное заболевание группы ферментопатий, связанное с нарушением метаболизма аминокислот, главным образом фенилаланина. Сопровождается накоплением фенилаланина и его токсических продуктов, что приводит к тяжёлому поражению ЦНС, проявляющемуся, в частности, в виде нарушения умственного развития.
Этиология. В большинстве случаев (классическая форма) заболевание связано с резким снижением или полным отсутствием активности печёночного фермента фенилаланин-4-гидроксилазы, который в норме катализирует превращение фенилаланина в тирозин. Заболевание наследуется по аутосомно-рецессивному типу.
Патогенез. Вследствие метаболического блока активируются побочные пути обмена фенилаланина, и в организме происходит накопление его токсичных производных — фенилпировиноградной и фениломолочной кислот, которые в норме практически не образуются. Кроме того, образуются также почти полностью отсутствующие в норме фенилэтиламин и ортофенилацетат, избыток которых вызывает нарушение метаболизма липидов в головном мозге. это и ведёт к прогрессирующему снижению интеллекта у таких больных вплоть доидиотии. Окончательно механизм развития нарушений функций мозга при фенилкетонурии остается неясным. Среди причин также предполагается дефицит нейромедиаторов мозга, вызванный относительным снижением количества тирозина и других «больших» аминокислот, конкурирующих с фенилаланином при переносе через гематоэнцефалический барьер, и прямое токсическое действие фенилаланина.
Алкапто́нури́я — наследственное заболевание, характеризующееся расстройством обмена тирозина и экскрецией с мочой большого количества гомогентизиновой кислоты.
Алкаптонурия возникает вследствие мутации гена, кодирующего синтез оксидазы гомогентезиновой кислоты. Данная патология характеризуется аутосомно-рецессивным типом наследования.
В нормальных условиях гомогентезиновая кислота — промежуточный продукт распада тирозина и фенилаланина — переводится в малеилацетоуксусную кислоту, из которой в конечном счёте образуются фумаровая и ацетоуксусная кислоты, вступающие в другие биохимические циклы. Из-за дефекта фермента этот процесс тормозится, и остающаяся в избытке гомогентезиновая кислота превращается полифенолоксидазой в хиноновый полифенол (алкаптон или бензохинонацетат), который и выводится почками. Не полностью экскретируемый мочой алкаптон откладывается в хрящевой и другой соединительной ткани, обусловливая их потемнение и повышенную хрупкость. Чаще всего вперёд появляется пигментация склер и ушных хрящей.
Альбинизм (лат. albus — белый) — врождённое отсутствие пигмента кожи, волос, радужной и пигментной оболочек глаза.Различают полный и частичный альбинизм.Наследуется по аутосомно- рецессивному типу или возникает в результате эндокринных расстройств и воздействий на развивающийся плод инфекций, интоксикаций и других неблагоприятных факторов. В основе альбинизма лежит нарушение образования пигмента меланина из тирозина.
3. Недостаточность внешнего дыхания, виды, общая этиология и патогенез.
источник
В основе многих заболеваний лежат нарушения функционирования ферментов в клетке — энзимопатии. Различают первичные (наследственные) и вторичные (приобретённые) энзимопатии. При первичных энзимопатиях дефектные ферменты наследуются, в основном, по аутосомнорецессивному типу. Гетерозиготы, чаще всего, не имеют фенотипических отклонений. Первичные энзимопатии обычно относят к метаболическим болезням, так как происходит нарушение определённых метаболических путей.
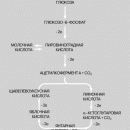
Фенилкетонурия (ФКУ). Фенилаланин (ФА) принадлежит к числу незаменимых аминокислот. Только часть ФА используется для синтеза белков; основное количество этой аминокислоты окисляется до тирозина. Реакцию гидроксилирования ФА катализирует фермент – фенилаланингидроксилаза. Причина ФКУ – недостаточность этого фермента. В результате этого нарушения ФА не превращается в тирозин, из которого образуется фенилпировиноградной кислоты (ФПВК), которая выделяется с мочой и потом, вследствие чего от больных детей исходит «мышиный» запах. Высокая концентрация ФПВК приводит к нарушению формирования миелиновой оболочки вокруг аксонов в ЦНС. ФПВК является нейротропным ядом, в результате чего повышаются возбудимость, тонус мышц, развиваются гиперрефлексия, тремор, судорожные эпилептиформные припадки. Позже присоединяются нарушения высшей нервной деятельности, умственная отсталость, микроцефалия. У больных детей наблюдается слабая пигментация из-за нарушения синтеза меланина. Диагностика заболевания осуществляется биохимическими методами: ещё до развития клинической картины в моче определяется ФПВК, в крови — высокое содержание фенилаланина. В родильных домах обязательно проводится скрининг-тест на фенилкетонурию.
Алкаптонурия. Нарушено окисление гомогентизиновой кислоты в тканях (гомогентизиновая кислота — промежуточный метаболит катаболизма тирозина). У таких больных наблюдают недостаточность фермента окисления гомогентизиновой кислоты — диоксигеназы гомогентизиновой кислоты, приводящей к развитию заболевания. В результате увеличиваются концентрация гомогентизиновой кислоты и выведение её с мочой. В присутствии кислорода гомогентизиновая кислота превращается в соединение чёрного цвета — алкаптон. Поэтому моча таких больных на воздухе окрашивается в чёрный цвет. Алкаптон также образуется и в биологических жидкостях, оседая в тканях, коже, сухожилиях, суставах. При значительных отложениях алкаптона в суставах нарушается их подвижность.
Альбинизм. При альбинизме нарушен синтез в меланоцитах пигментов — меланинов. Меланин находится в коже, волосах, радужке, пигментном эпителии сетчатки глаза и влияет на их окраску. При альбинизме наблюдают слабую пигментацию кожи, светлые волосы, красноватый цвет радужки глаза из-за просвечивающих капилляров. Проявление альбинизма связано с недостаточностью фермента тирозингидроксилазы (тирозиназы) — одного из ферментов, катализирующего метаболический путь образования меланинов.
Болезнь Хартнупа. Причина — недостаточность белков-переносчиков триптофана в кишечной стенке и почечных канальцах. Наблюдаются:повышенное образование индикана, который превращается в индиго синего цвета – симптом «голубых пеленок»; аминоацидурия; признаки пеллагры. нарушение психики. Лечение: полноценная белковая диета, прием никотинамида, предохранение от УФО.
ТВОРЧЕСКАЯ РАБОТА СТУДЕНТОВ. Тема реферативного сообщения/презентации —«Энзимопатии обмена аминокислот. Генетический скрининг новорожденных».
Лекция № 3. Конечные продукты обмена простых белков.
План лекции:
1. Реакции образования аммиака в организме. Токсичность аммиака.
2. Способы обезвреживание аммиака в организме. Орнитиновый цикл. Диагностическое значение определения мочевины.
3. Обмен креатина. Синтез креатина, образование креатинфосфата и креатинина. Диагностическое значение определения креатина, креатинина и креатинкиназы.
Содержание лекционного материала:
1. Реакции образования аммиака в организме. Токсичность аммиака.
Источники аммиака в организме:
a) Дезаминирование аминокислот.
b) Дезаминирование биогенных аминов.
c) Дезаминирование пуриновых и пиримидиновых азотистых оснований.
d) Гниение аминокислот в кишечнике.
Катаболизм аминокислот в тканях происходит постоянно со скоростью ∼100 г/сут. При этом в результате дезаминирования аминокислот освобождается большое количество аммиака. Значительно меньшие количества его образуются при дезаминировании биогенных аминов и нуклеотидов.
Часть аммиака образуется в кишечникев результате действия бактерий на пищевые белки (гниение белков вкишечнике) и поступает в кровь воротной вены. Концентрация аммиака в крови воротной вены существенно больше, чем в общем кровотоке. В печени задерживается большое количество аммиака, что поддерживает низкое содержание его в крови. Концентрация аммиака в крови в норме редко превышает 25-40 мкмоль/л.
Аммиак — токсичное соединение. Даже небольшое повышение его концентрации оказывает неблагоприятное действие на организм, и прежде всего на ЦНС. Так, повышение концентрации аммиака в мозге до 0,6 ммоль вызывает судороги. К симптомам гипераммониемии относят тремор, нечленораздельную речь, тошноту, рвоту, головокружение, судорожные припадки, потерю сознания. В тяжёлых случаях развивается кома с летальным исходом.
2. Способы обезвреживание аммиака в организме.
2.1. Локальный – в тканях. Основной реакцией связывания аммиака, протекающей во всех тканях организма, является синтез глутамина под действием глутаминсинтетазы:
Глутамин легко транспортируется через клеточные мембраны путём облегчённой диффузии (для глутамата возможен только активный транспорт) и поступает из тканей в кровь. Основными тканями-поставщиками глутамина служат мышцы, мозг и печень. С током крови глутамин транспортируется в почки и печень.
В мозге и некоторых других органах может протекать восстановительное аминирование α-кетоглутаратапод действием глутаматдегидрогеназы, катализирующей обратимую реакцию. Однако этот путь обезвреживания аммиака в тканях используется слабо, так как глутаматдегидрогеназа катализирует преимущественно реакцию дезаминирования глутамата.
2.2. 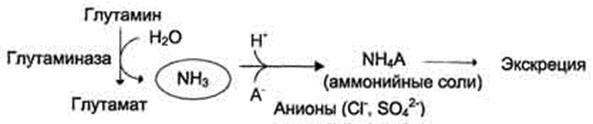 Образование аммонийных солей. В почкахпроисходит гидролиз глутамина под действием глутаминазы с образованием аммиака. Образующийся аммиак нейтрализует кислые продукты обмена и в виде аммонийных солей, которые экскретируются с мочой:
Образование аммонийных солей. В почкахпроисходит гидролиз глутамина под действием глутаминазы с образованием аммиака. Образующийся аммиак нейтрализует кислые продукты обмена и в виде аммонийных солей, которые экскретируются с мочой:
2.3. Орнитиновый цикл. Диагностическое значение определения мочевины.
Мочевина — основной конечный продукт азотистого обмена,в составе которого из организма выделяется до 90% всего выводимого азота. В норме концентрация мочевины в сыворотке/плазме крови – 2,61-8,35 ммоль/л (уреазный метод).
Мочевина выводится из организма с мочой. Азот мочевины составляет 85-90% от общего азота мочи. В норме у взрослых выводится мочевины с мочой 20-35 г/сут (330-580 ммоль/сут).
При повышении количества потребляемых с пищей белков экскреция мочевины увеличивается. Мочевина синтезируется только в печени. В 40-х годах XX века немецкие биохимики Г. Кребс и К. Гензелейт установили, что синтез мочевины представляет собой циклический процесс, состоящий из нескольких стадий, ключевым соединением которого, замыкающим цикл, является орнитин. Поэтому процесс синтеза мочевины получил название «орнитиновый цикл»,или «цикл Кребса-Гензелейта».
Суммарное уравнение синтеза мочевины:
NH3 + Аспартат + СО2 + 3 АТФ + 2 Н2О → Мочевина + Фумарат + 2 АДФ +
В первой реакции процесса аммиак связывается с СО2 с образованием карбамоилфосфата, при этом затрачиваются 2 молекулы АТФ. Реакция происходит в митохондриях гепатоцитов под действием фермента карбамоилфосфатсинтетазы I. Карбамоилфосфат затем включается в орнитиновый цикл и используется для синтеза мочевины.
Биологическая роль орнитинового цикла
Орнитиновый цикл в печени выполняет 2 функции:
· превращение азота аминокислот в мочевину, которая экскретируется и предотвращает накопление токсичных продуктов, главным образом аммиака;
· синтез аргинина и пополнение его фонда в организме.
Нарушение реакций обезвреживания аммиака может вызвать повышение содержания аммиака в крови— гипераммониемию, что оказывает токсическое действие на организм. Причинами гипераммониемии могут выступать как генетический дефект ферментов орнитинового цикла в печени, так и вторичное поражение печени в результате цирроза, гепатита и других заболеваний.
Все нарушения орнитинового цикла приводят к значительному повышению в крови концентрации аммиака, глутамина и аланина.
Гипераммониемиясопровождается появлением следующих симптомов:
- тошнота, повторяющаяся рвота;
- головокружение, судороги;
- потеря сознания, отёк мозга (в тяжёлых случаях);
- отставание умственного развития (при хронической врождённой форме).
Клинико-диагностическое значение исследования концентрации мочевины в сыворотке крови и моче.
Определение концентрации мочевины в сыворотке крови используется для оценки функции печени и выделительной функции почек. Критическая концентрация мочевины в крови: ˃28,0 ммоль/л. Причины увеличения концентрации мочевины в сыворотке крови:острые или хронические заболевания почек; обтурация мочевыводящих путей; снижение тока крови через почечные клубочки (застойная сердечная недостаточность, обширные ожоги, тяжелая диарея и рвота, инфаркт миокарда и др.); повышенный катаболизм белков (длительное голодание, кровотечения в ЖКТ, инфекции, канцерогенез, сахарный диабет и др.); снижение концентрации Cl — . Причины снижения концентрации мочевины в сыворотке крови: функциональная недостаточность печени (гепатит, цирроз, отравления мышьяком и др.); энзимопатии синтеза мочевины; повышенная скорость клубочковой фильтрации, обусловленная беременностью, чрезмерным объемом внутривенных инфузий, неадекватной секрецией антидиуретического гормона; мальабсорбция (целиакия). Влияющие факторы. Увеличение концентрации: диета с высоким содержанием белка, прием нефротоксичных лекарств, повышенный распад тканей при гипертиреозе, приеме глюкокортикоидов. Уменьшение концентрации:вегетарианская диета.
Определение концентрации мочевины в моче позволяет контролировать азотистый баланс у больных в тяжелом состоянии, получающих зондовое энтеральное и парентеральное питание. Корректно оценить азотистый баланс с использованием этого теста можно лишь при отсутствии тяжелой печеночной или почечной патологии. Увеличением концентрации мочевины в моче, указывающим на отрицательный азотистый баланс, сопровождаются: послеоперационный период, лихорадка; гипертиреоз; тяжелые острые или хронические соматические заболевания. Уменьшением, указывающим на положительный азотистый баланс, сопровождаются: беременность; период роста у детей; период восстановления после тяжелых заболеваний.
3. Обмен креатина. Синтез креатина, образование креатинфосфата и креатинина. Диагностическое значение определения креатина, креатинина и креатинкиназы.
Синтез креатина. Креатин необходим для образования в мышцах высокоэнергетического соединения — креатинфосфата. Синтез креатина идёт в 2 стадии с участием 3-х аминокислот: аргинина, глицина и метионина. В почкахобразуется гуанидинацетат при действии глицинамидинотрансферазы.Затем гуанидинацетат транспортируется в печень,где происходит реакция его метилирования:
SAM – S-аденозилметионин – активная форма метионина – донор СН3-группы.
 Креатин с кровотоком переносится в мышцы и клетки мозга,где из него образуется высокоэнергетическое соединение — креатинфосфат.
Креатин с кровотоком переносится в мышцы и клетки мозга,где из него образуется высокоэнергетическое соединение — креатинфосфат.
 |
Эта реакция легко обратима и катализируется ферментом креатинкиназой. Обнаружено три изоферментные формы креатинкиназы (КК-ВВ – мозговой тип, КК-МВ – сердечный тип, КК-ММ – мышечный тип). Измерение активности общей креатинкиназы и ее изоферментов используется в лабораторной диагностике.
Креатинфосфат играет важную роль в обеспечении энергией работающей мышцы в начальный период.
Креатинин– это конечный продукт неферментативного превращения креатина и креатинфосфата, участвующих в энергообеспечении мышечного сокращения:
Креатинин выводится из организма через почки с мочой, относится к беспороговым веществам, которые поступают в мочу путем фильтрации и не реабсорбируются в канальцах. Нормы креатинина в сыворотке крови у взрослых: мужчины – 40-100 мкмоль/л, женщины – 44-88 мкмоль/л; креатинина в моче у взрослых — 4,4-17,7 ммоль/сут.
Клинико-диагностическое значение исследования концентрации креатина и креатинина в сыворотке крови и моче. Гиперкреатининемия – увеличение уровня креатинина в крови обусловлено как усиленным образованием, так и задержкой его в организме. Критическая концентрация креатинина в сыворотке крови: ˃400 мкмоль/л. Ретенционная гиперкреатининемия обусловлена нарушением (острым и хроническим) функции почек любого происхождения. Продукционная гиперкреатининемия отмечается при кишечной непроходимости, декомпенсации деятельности ССС, пневмонии, лихорадочных состояниях, гипертиреозе, голодании, усиленной мышечной работе. Снижение уровня креатинина в сыворотке крови коррелирует с уменьшением мышечной массы (мышечные дистрофии и атрофии, параличи и др.). Увеличение суточного выведения креатинина с мочойуказывает на увеличение почечной фильтрации и/или повышенное образование креатинина в скелетных мышцах. Увеличением концентрации сопровождаются: усиленная мышечная работа, лихорадочные состояния, пневмония, акромегалия, сахарный диабет, острые инфекционные заболевания. Уменьшение выведения креатинина с мочой наблюдается при мышечной атрофии, параличах, хронических заболеваниях почек, острой почечной недостаточности, голодании. В моче помимо эндогенного креатинина содержится экзогенный креатинин, поступающий в организм с мясной пищей. На концентрацию креатинина в крови и моче влияет характер диеты: преимущественно мясная диета приводит к увеличению, и наоборот, вегетарианская диета – к снижению концентрации креатинина в крови и моче.
Креатинемия (креатин + греч. haima кровь) — наличие в крови креатина. В плазме взрослого человека концентрации креатина в норме составляет 15,25—76,25 мкмоль/л. Повышение уровня креатина в крови (гиперкреатинемия) может быть при избыточном употреблении мяса, а также при заболеваниях мышечной системы, при кишечной непроходимости. поражениях печени. Креатинв моче взрослых людей в норме практически отсутствует. В первые годы жизни ребенка возможна «физиологическая креатинурия». По-видимому, появление креатина в моче детей раннего возраста обусловлено усиленным синтезом креатина, опережающим развитие мускулатуры. Некоторые исследователи к физиологическим явлениям относят и креатинурию стариков, которая возникает как следствие атрофии мышц и неполного использования образующегося в печени креатина. При концентрации креатина в плазме крови свыше 122,0 мкмоль/л он начинает выделяться с мочой. Наибольшее содержание креатина в моче наблюдается при патологических состояниях мышечной системы и прежде всего при миопатии, или прогрессирующей мышечной дистрофии. Принято считать, что креатин в моче (креатинурия) больных миопатией может появляться в результате нарушения в скелетной мускулатуре процессов его фиксации (удержания) и фосфорилирования. Если нарушен процесс синтеза фосфокреатина, то не образуется и креатинин; содержание последнего в моче резко снижается. В результате креатинурии и нарушения синтеза креатинина резко повышается креатиновый показатель мочи: (количество креатина + количество креатинина)/(количество креатинина). В норме этот показатель близок к 1,1.
ТВОРЧЕСКАЯ РАБОТА СТУДЕНТОВ. Тема реферативного сообщения презентации«Энзимопатии цикла синтеза мочевины. Лабораторная диагностика гипераммониемии».
Дата добавления: 2015-10-01 ; просмотров: 1549 | Нарушение авторских прав
источник
Наследственные нарушения обмена аминокислот (первичные аминоацидопатии)— это большая группа врожденных заболеваний, в основе которых лежит генетически обусловленное нарушение синтеза различных ферментов, а основным клиническим проявлением — является метаболическая энцефалопатия. Заболевания этой группы моногенного происхождения и имеют аутосомно-рецессивный тип наследования.
Фенилкетонурия (ФКУ)– наследственная задержка психического развития, обусловленная недостаточностью фермента – фенилаланингидроксилазы, необходимой для превращения аминокислоты фенилаланина в тирозин. Накапливающийся фенилаланин (ФА) оказывает токсическое действие на ткань головного мозга.
Болезнь впервые была описана в 1934 А.Фёллингом. Эрвис в 1947 году определил патологический фермент – фенилаланингидроксилазу, который берет участие в гидроксилировании фенилаланина в тирозин. Бикел в 1953 году показал, что ограничение в рационе ФА улучшает психическое развитие.
Частота случаев фенилкетонуриисоставляет1:6000 – 1:10 000 новорожденных, независимо от пола и наиболее распространена среди европейцев. Выявлен Мутантный ген, который отвечает за синтез фермента фенилаланингидроксилазы.
Фенилаланин (ФА) принадлежит к числу незаменимых аминокислот. Для синтеза белков используется только часть ФА, а основное количество этой аминокислоты окисляется до тирозина. Если фермент фенилаланингидроксилаза не активен, то ФА не превращается в тирозин, а накапливается в сыворотке крови в больших количествах в виде фенилпировиноградной кислоты (ФПВК), которая выделяется с мочой и потом, вследствие чего от больных исходит характерный «мышиный» запах. Высокая концентрация ФПВК приводит к нарушению формирования миелиновой оболочки вокруг аксонов в ЦНС.
Дети с фенилкетонурией рождаются здоровыми, но в первые же недели жизни у них развиваются клинические проявления заболевания – тетрада признаков:
1.Задержка психического развития.
3.Склонность к развитию дерматита.
4.Нарушение пигментного обмена. Дети с ФКУ с бледной кожей, голубыми глазами, в результате снижения меланина в коже.
ü От больных с ФКУ исходит специфический «мышиный» запах.
Епилептиформные припадки возникают в виде кивательных, тонических и полиморфных судорог, закатывания глаз, вздрагивания. Такие клинические проявления обусловлены нейротропным влиянием ФПВК, в результате чего повышаются возбудимость, тонус мышц, развиваются гиперрефлексия, тремор, а также судорожные припадки. В межприступный период ребенок вялый, отмечается мышечная гипотония. К 3-5 месяцам больной проявляет полное безразличие к окружающим и не узнает родителей. Умственная отсталость достигает степени имбецильности или идиотии, что ведет к инвалидизации.
В настоящее время выделяют 5 нозологических типов фенилкетонурии, обусловленные мутацией различных звеньев биосонтеза кофактора фенилаланингидроксилазы:
· тип I — классическая ФКУ, при которой фермент фенилаланингидроксилаза отсутствует, в крови определяется повышенное содержание фенилаланина, в моче — фенилаланин и его метаболиты (фенилпируват, фениллактат, О-гидроксифенилацетат);
· тип II — вариантная ФКУ, наблюдается дефект фенилаланингидроксилазы, в крови — повышенное содержание фенилаланина, без метаболитов в моче;
· тип III — транзиторная неонатальная ФКУ с дефектом фенилаланингидроксилазы, избытком фенилаланина в крови, без метаболитов в моче;
· тип IV — при котором отсутствует фермент дигидроптеринредуктаза, в крови повышено содержание фенилаланина, в моче — вариабельные метаболиты;
· тип V — связанный с генетическим дефектом синтеза биоптерина, в крови — избыток фенилаланина, в моче — вариабельные метаболиты.
Каждая форма ФКУ характеризуется умственной отсталостью и тяжелой неврологической симптоматикой. Вариантные формы фенилкетонурии резистентны к диетотерапии, которая является основой лечения классической ФКУ.
Лечение: проводится диетотерапией — необходимо придерживаться диеты со строгими ограничениями содержания в продуктах фенилаланина из-за того, что эта аминокислота в огромном количестве есть в белке, из рациона абсолютно исключаются вся белковая пища животного происхождения — это, молоко, мясо, рыба, грибы и прочее.
Для ранней диагностики (на доклиническом уровне) фенилкетонурии разработаны и внедрены в практику программы массового обследования новорожденных в родильных домах. Они основаны на выявлении повышенного уровня фенил-аланина в образцах крови, собранных на 5-6 днях жизни.
Алкаптонурия
Алкаптонурия [alcaptonuria; алкаптон (гомогентизиновая кислота) + греч. uron моча] — врожденное отсутствие фермента — оксидазы гомогентизиновой кислоты, играющего существенную роль в процессе нормального расщепления аминокислот тирозина и фенилаланина. При этом обмен тирозина достигает лишь стадии гомогентизиновой кислоты, которая из-за врожденного недостатка фермента — кислой оксидазы, расщепляющего ее бензольное ядро, дальнейшей трансформации не подвергается и откладывается в коже, костях, хрящах, склере и в других органах, а также выделяется с мочой. В результате окисления гомогентизиновая кислота превращается в меланиноподобный пигмент неизвестной структуры. Аккумуляция кислоты и отложения пигмента в хрящах и других соединительнотканных структурах определяют развитие симптомокомплекса, известного как охроноз: темно-коричневый оттенок кожи и глаз, а также приводит к прогрессирующему поражению суставов, особенно позвоночника.
Впервые заболевание описал Scribonius в 1584 году. В 1859 году C.H.Boedeker назвал алкаптоном вещество, содержащееся в моче и обусловливающее потемнение мочи при соприкосновении с воздухом в результате окисления гомогетензиновой кислоты, что приводит к образованию темного пигмента – алкаптона.
Клиническая картина. Алкаптонурию можно легко распознать еще в младенческом возрасте из-за характерного окрашивания пленок. На пеленках, смоченных мочой ребенка, остаются темные пятна, а при отстаивании она быстро чернеет, вследствие окисления содержащейся в ней гомогетензиновой кислоты. Гомогетинзиновая кислота, откладывающаяся в тканях, служит исходным материалом для образования пигмента, вследствие чего кожа окрашивается в желтый цвет (охроноз). Кожные проявления характеризуются развитием выраженной пигментации над скуловой частью лица, в подмышечных впадинах и на гениталиях. Отложения пигмента можно обнаружить также в склере. Иногда пигментация принимает распространенный характер. Больные отмечают общую слабость. В более поздние сроки пигмент откладывается в хрящах, почках, надпочечниках, щитовидной, поджелудочной, предстательной железах, придатках яичка. Может наблюдаться темное окрашивание склер и хрящей, близко расположенных к коже (ушных раковин, крыльев носа). Отложение пигмента наблюдается и в сердечной мышце, клапанах сердца, эндотелии сосудов. В связи с поражением хрящей суставовпосле 20-30 лет жизни и позже развивается их деформация. Чаще поражаются большие суставы — плечевые, коленные, а также позвоночник.
Лечение алкаптонурии симптоматическое: назначается диета с низким содержанием белка. Прогноз заболевания для жизни благоприятный
Аминоацидурия (aminoaciduria; англ. amino acid аминокислота + греч. uron моча; син. гипераминоацидурия) — повышенное выведение с мочой одной или нескольких аминокислот или наличие в моче продуктов их обмена, в норме не содержащихся в ней.
Клинически: атрофия сосудистой оболочки глаза и сетчатки, отставание в умственном развитии.
Лечение проводят индивидуально (в зависимости от конкретного дефекта). Общие принципы:
Диета с пониженным содержанием белка
Последнее изменение этой страницы: 2017-01-24; Нарушение авторского права страницы
источник
Причина заболевания — дефект диоксигеназы гомогентизиновой кислоты. Для этой болезни характерно выделение с мочой большого количества гомогентизиновой кислоты, которая, окисляясь кислородом воздуха, образует тёмные пигменты алкаптоны. Это метаболическое нарушение было описано ещё в XVI веке, а само заболевание охарактеризовано в 1859 г. Клиническими проявлениями болезни, кроме потемнения мочи на воздухе, являются пигментация соединительной ткани (охроноз) и артрит. Частота — 2-5 случаев на 1 млн новорождённых. Заболевание наследуется по аутосомнорецессивному типу. Диагностических методов выявления гетерозиготных носителей дефектного гена к настоящему времени не найдено.
Причина метаболического нарушения — врождённый дефект тирозиназы. Этот фермент катализирует превращение тирозина в ДОФА в меланоцитах. В результате дефекта тирозиназы нарушается синтез пигментов меланинов.
Клиническое проявление альбинизма (от лат. albus — белый) — отсутствие пигментации кожи и волос. У больных часто снижена острота зрения, возникает светобоязнь. Длительное пребывание таких больных под открытым солнцем приводит к раку кожи. Частота заболевания 1:20 000.
Нарушение синтеза катехоламинов может вызывать различные нервно-психические заболевания, причём патологические отклонения наблюдаются как при снижении, так и при увеличении их количества.
Заболевание развивается при недостаточности дофамина в чёрной субстанции мозга. Это одно из самых распространённых неврологических заболеваний. При этой патологии снижена активность тирозингидроксилазы, ДОФА-декарбоксилазы. Заболевание сопровождается тремя основными симптомами: акинезия (скованность движений), ригидность (напряжение мышц), тремор (непроизвольное дрожание). Дофамин не проникает через гематоэнцефалический барьер и как лекарственный препарат не используется. Для лечения паркинсонизма предлагаются следующие принципы:
заместительная терапия препаратами-предшественниками дофамина (производными ДОФА) — леводопа, мадопар, наком и др.
подавление инактивации дофамина ингибиторами МАО (депренил, ниаламид, пиразидол и др.).
Депрессивные состояния часто связаны со снижением в нервных клетках содержания дофамина и норадреналина.
Гиперсекреция дофамина в височной доле мозга наблюдается при шизофрении.
Конечные продукты обмена белков:
C, H, O, N , S. – CO2 , H2O, NH3, H2S.
Соли аммония выводятся с мочой:
В почках также происходит гидролиз глутамина под действием глутаминазы с образованием аммиака. Этот процесс является одним из механизмов регуляции кислотно щелочного равновесия в организме и сохранения важнейших катионов для поддержания осмотического давления. Глутаминаза почек значительно индуцируется при ацидозе, образующийся аммиак нейтрализует кислые продукты обмена и в виде аммонийных солей экскретируется с мочой .
Эта реакция защищает организм от излишней потери ионов Na+ и К+, которые также могут использоваться для выведения анионов и утрачиваться. При алкалозе количество глутаминазы в почках снижается.
В почках образуется и выводится около 0,5 г солей аммония в сутки.
В печени – синтез мочевины ( в следующих вопросах будет подробно описано. Честно говоря, странный вопрос – скорее всего надо просто сказать где образуется.)
Вопрос №38. Основные источники и пути обезвреживания аммиака в организме.
Непрямое дезаминирование(основной путь дезаминирования амк)
Окислителное дезаминирование глутамата
Окислителное дезаминирование амк(малозначимый путь дезаминирования)
неокислительное дезаминирование Гис,Сер ,Тре
окислительное дезаминирование (путь инактивации биогенных аминов)
гидролитическое дезаминирование АМФ
гниение белков в кишечнике в результате действия бактерий на пищевые белки.
Пути обезвреживания:связывание аммиака с образованием нетоксичных соединений,которые выводятся из организма вместе с мочой
Синтез глутамина под действием глутаматсинтетазы
Синтез аспарагина под действием аспарагинсинтета
Синтез мочевиы в печени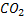
 +
+
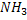 +
+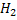
 O
O
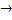 карбомоилфосфат – 2АТФ
карбомоилфосфат – 2АТФ
Восстановительное аминирование а-кетоглутарата
Вопрос №39 Роль глутамина в обезвреживании и транспорте аммиака в организме.
глутамин образуется при обезвреживании аммиака (мышцы, мозг, печень)Связывание аммиака глутамином протекает во всех тканях организма
Глутамин легко транспортируется через клеточные мембраны путём облегчённой диффузии и транспортируется из тканей в кровь.
Вопрос №40 Глутамин как донор амидной группы при синтезе ряда соединений.
Высокий уровеньглутамина в крови и легкость его поступления в клетки обусловливают использования глутамина во многих анаболических процессах
Глутамин — основной донор азота в организме. Амидный азот глутамина используется для синтеза пуриновых и пиримидиновых нуклеотидов, аспарагина, аминосахаров и других соединений
Вопрос №41 Синтез мочевины, химизм, ферменты, энергетика, происхождение атомов азота в мочевине
В реакциях орнитинового цикла расходуются четыре макроэргических связи трёх молекул АТФ на каждый оборот цикла.Источник первого азота-аммиак. Аспартат — источник второго атома азота мочевины
Вопрос №42 Связь орнитинового цикла с циклом трикарбоновых кислот.
Взаимосвязь орнитинового цикла и общего пути катаболизма. Фумарат, образующийся в результате расщепления аргининосукцината, превращается в малат, который затем переносится в митохондрии, включается в ЦТК и дегидрируется с образованием оксалоацетата. Эта реакция сопровождается выделением 3 молекул АТФ, которые и компенсируют затраты энергии на синтез одной молекулы мочевины. ЦЦ
ЦТК и орнитиновый цикл протекают в печени.
Вопрос№43 Нарушение синтеза и выведения мочевины. Гипераммониемия, происхождение
Нарушение реакций обезвреживания аммиака может вызвать повышение содержания аммиака в крови — гипераммониемию, что оказывает токсическое действие на организм. Причинами гипераммониемии могут выступать как генетический дефект ферментов орнитинового цикла в печени, так и вторичное поражение печени в результате цирроза, гепатита и других заболеваний. Известны пять наследственных заболеваний, обусловленных дефектом пяти ферментов орнитинового цикла
Наследственные нарушения орнитинового цикла и основные их проявления
Карбамоил- фосфат- синтетаза I
В течение 24-48 ч после рождения кома, смерть
Орнитин- карбамоил- трансфераза
Гипотония, снижение толерантности к белкам
Аргинино- сукцинат- синтетаза
Гипераммониемия тяжёлая у новорождённых. У взрослых — после белковой нагрузки
Гипераммонимия, атаксия, судороги, выпадение волос
Аргини- носукци- нат, Глн, Ала, Лиз
Основной диагностический признак — повышение концентрации аммиака в крови. Содержание аммиака в крови может достигать 6000 мкмоль/л (в норме — 60 мкмоль/л). Однако в большинстве хронических случаев уровень аммиака может повышаться только после белковой нагрузки или в течение острых осложнённых заболеваний.
Вопрос№44 Определение мочевины в сыворотке крови, принцип метода, диагностическое значение. Количество морчевины в сыворотке крови определяется уреазным методом
Под действием уреазы мочевина распадается на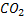
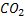 и
и
 ,которые в щелочной среде с гипохлоритом натрия и фенолом образуют нофенол синего цвета(метод Бертло).Светопоглощение образующегося продукта пропорционально содержанию мочевины в образце.
,которые в щелочной среде с гипохлоритом натрия и фенолом образуют нофенол синего цвета(метод Бертло).Светопоглощение образующегося продукта пропорционально содержанию мочевины в образце.
Значение:при повышении концентрации мочевины , которое встречается при хронических поражениях почек, усиленном распаде белков в тканях, непроходимости кишечника ,закупорке мочевыводящих путей.Снижение уровня мочевины наблюдается при голодании, безбелковой диете, ферментных дефектах мочеобразования.
45. Образование и выведение солей аммония. Глутаминаза почек
Почка располагает рядом ферментных систем, разрушающих глутамин, но, подобно большинству реакций анаболизма аминокислот, синтез глутамина не осуществляется. Глутамин доставляется к почкам током крови. Клетки почек жадно поглощают из циркулирующей крови глутамин, который образуется в основном за счет аммиака клеток печени. В почках же аммиак образуется преимущественно из глутамина, так что последний можно рассматривать как нетоксичную форму аммиака, который совершает челночные движения между печенью и почками. Глутаминаза почек действует подобно глутаминазе печени, освобождая глутамат и NH3 путем простой реакции гидролиза Однако фермент почек отличается тем, что его активность значительно возрастает под влиянием неорганического фосфата. Образующийся глутамат может, конечно, подвергаться затем дезаминированию при участии глутаматдегидрогеназы, так что из глутамина в конечном итоге образуются две молекулы аммиака.
В общем регулирует кислотно-щелочное равновесие в организме и задерживает натрий и калий.
источник
Нарушение обмена фенилаланина и тирозина
В печени здоровых людей небольшая часть фенилаланина (до 10%) превращается в фениллактат и фенилацетилглутамин. Этот путь катаболизма фенилаланина становится главным при нарушении основного пути – превращения в тирозин, катализируемого фенилаланингидроксилазой. Такое нарушение сопровождается гиперфенилаланинемией и повышением в крови и моче содержания фенилаланина и его метаболитов альтернативного пути. Классическая фенилкетонурия – наследственное заболевание, связанное с мутациями в гене фенилаланингидроксилазы. Наиболее тяжелые проявления фенилкетонурии – нарушение умственного и физического развития, судорожный синдром, нарушение пигментации. Эти проявления связаны с токсическим действием на клетку высоких концентраций фенилаланина, фенилпирувата, фениллактата.
Тирозинемии
Наследственные нарушения метаболизма тирозина в печени. Известно два типа.
I тип – дефект фермента фумарилацетоацетатгидроксилазы, из-за которого накапливаются в крови токсические метаболиты что приводит к тяжелому поражению печени и почек.
При II типе нет фермента тирозинаминотрансферазы. Повышется концентрация тирозина, наблюдается гиперкератоз ладоней и подошв.
Алкаптонурия
Причина заболевания – дефект диоксигеназы гомогентизиновой кислоты. Для этой болезни характерно выделение с мочой большого количества гомогентизиновой кислоты, которая, окисляясь воздухом, образует темные пигменты алкаптоны. Кроме того наблюдается пигментация соединительной ткани. Умственное и физическое развитие не нарушено.
Обусловлен отсутствием тирозиназы и, соответственно, нарушается синтез пигментов меланинов. Клиническое проявление – отсутствие пигментации кожи и волос. Умственное развитие не страдает. У людей с альбинизмом повышенная склонность к солнечным ожогам.
ГЛАВА 26
обмЕн нуклеотидов
Практически все клетки организма способны к синтезу нуклеотидов (исключение составляют некоторые клетки крови). Другим источником этих молекул могут быть нуклеиновые кислоты собственных тканей и пищи, однако эти источники имеют лишь второстепенное, вспомогательное значение.
Пуриновые и пиримидиновые нуклеотиды являются существенными компонентами клеток. Они или их производные выполняют различные функции:
· Нуклеозидтрифосфаты (НТФ) используются в качестве субстратов синтеза ДНК и РНК, без которых невозможны образование белков и клеточная пролиферация.
· Природа выбрала цикл АДФ-АТФ в качестве универсального механизма трансформации энергии окисления в энергию биосинтетических процессов. В некоторых биологических процессах и другие НТФ используются в качестве источника энергии.
· Производные нуклеотидов служат донорами активных субстратов в синтезе гомо- и гетерополисахаридов, липидов и белков. Например: УДФ-глюкоза, УДФ-галактоза, ГДФ-манноза, УДФ-N-ацетилглюкозамин или ЦМФ-ацетилнейраминовая кислота принимают участие в синтезе гликогена и гликозаминогликанов; ЦДФ-холин – в синтезе фосфолипидов.
· УДФ-глюкуроновая кислота, ФАФС, S-аденозилметионин – наиболее частые участники универсальной системы детоксикации, обеспечивающей последующее выведение ксенобиотиков (чужеродных веществ) и некоторых собственных метаболитов из организма.
· АМФ входит в состав коферментов дегидрогеназ (НАД + , НАДФ + , ФАД, ФМН) и ацилирования (КоА).
· С помощью циклических форм нуклеотидов (цАМФ, цГМФ) осуществляется передача в клетку сигналов гормонов, факторов роста, нейромедиаторов и некоторых других регуляторных молекул.
Не нашли то, что искали? Воспользуйтесь поиском:
Лучшие изречения: Для студента самое главное не сдать экзамен, а вовремя вспомнить про него. 9756 —  | 7376 —
| 7376 —  или читать все.
или читать все.
193.124.117.139 © studopedia.ru Не является автором материалов, которые размещены. Но предоставляет возможность бесплатного использования. Есть нарушение авторского права? Напишите нам | Обратная связь.
Отключите adBlock!
и обновите страницу (F5)
очень нужно
источник
ОБМЕНА ВЕЩЕСТВ НАРУШЕНИЯ, возможные при любом заболевании нарушения каких-либо из многочисленных химических процессов, участвующих в обмене веществ в организме. Разнообразные патологические проявления включают изменения скорости роста, теплопродукции, выработки энергии для мышечной деятельности и энергетического обеспечения жизненно важных функций организма. Известно, однако, большое число т.н. болезней обмена веществ, или болезней метаболизма, причиной которых служит специфическое его нарушение; ниже описаны лишь наиболее важные из них.
Это врожденное нарушение обмена веществ, характеризующееся накоплением избыточного количества гликогена в тканях организма. Оно связано с недостаточностью фермента глюкозо-6-фосфатазы, который необходим для распада гликогена, в силу чего тот накапливается в тканях. Болезнь обычно проявляется уже в младенчестве отставанием в росте, выпячиванием живота из-за увеличения размеров печени и снижением уровня сахара в крови. Единственное средство лечения – диета; рекомендуется частое кормление и добавление в рацион глюкозы. С возрастом состояние ребенка постепенно улучшается.
– наследственная задержка психического развития, обусловленная недостаточностью единственного фермента – фенилаланингидроксилазы, необходимой для превращения аминокислоты фенилаланина в другую аминокислоту – тирозин. Накапливающийся фенилаланин оказывает токсическое действие на ткань головного мозга. Болезнь впервые была описана в 1934 А.Фёллингом. Она встречается с частотой 1 на 20 000 новорожденных независимо от пола и наиболее распространена среди европейцев.
Новорожденные внешне выглядят здоровыми, но в возрасте трех-четырех месяцев у них начинает проявляться отставание в психическом развитии. К 2–3 годам дети прекрасно развиваются физически, но психически отстают. Поскольку нарушение психического развития поддается лечению, крайне важна ранняя диагностика; в отсутствие лечения коэффициент интеллектуального развития (IQ) за каждые 10 недель снижается на 5 пунктов. Фенилкетонурию можно обнаружить уже в первый день жизни по результатам анализа крови или мочи новорожденного.
Единственным способом лечения является диета. Поскольку все обычные белковые продукты содержат фенилаланин (в количестве 4–6%), необходимо использовать лишенные этой аминокислоты синтетические продукты.
При нормальном метаболизме фенилаланина и тирозина (обе аминокислоты связаны между собой в обмене) образуется кожный черный пигмент меланин. Врожденное отсутствие этого пигмента в глазах, коже и волосах у лиц с альбинизмом обусловлено недостаточностью одного из ферментов метаболизма фенилаланина и тирозина.
Это заболевание вызывается генетически обусловленной недостаточностью фермента, участвующего в метаболизме гомогентизиновой кислоты – промежуточного продукта обмена фенилаланина и тирозина. Накапливающаяся гомогентизиновая кислота выделяется с мочой, придавая ей черный или коричневый цвет. В более позднем возрасте в соединительной ткани и хрящах откладывается синевато-черный пигмент и развивается артрит. В качестве лечения назначают диету, исключающую потребление фенилаланина и тирозина.
Неспособность организма разрушать холестерин и липопротеины низкой плотности (в составе которых он в основном находится) приводит к накоплению холестерина в тканях из-за чрезмерно высокого его уровня в крови. Состояние, при котором холестерин откладывается в подкожных тканях, называют ксантоматозом. Отложения холестерина в стенках кровеносных сосудов вызывают атеросклероз. При гиперхолестеринемии возможно также увеличение селезенки, печени или лимфатических узлов. Для лечения и профилактики используют диету.
Подагра и подагрический артрит – хронические заболевания, вызываемые нарушением обмена эндогенной (образующейся в организме) мочевой кислоты; ее соли (ураты) откладываются главным образом в хрящах, особенно суставных, и в почках, обусловливая болезненные воспалительные отеки. Накопление уратов можно предотвратить с помощью диеты. Для ослабления болей применяют специальные средства.
Многие процессы метаболизма непосредственно контролируются гормонами. Поэтому дисфункция эндокринных желез также может приводить к нарушениям обмена веществ. См. также ГОРМОНЫ; ДИАБЕТ САХАРНЫЙ; ЗОБ; НАСЛЕДСТВЕННОСТЬ; ПОДАГРА; ФЕНИЛКЕТОНУРИЯ.
источник