В основе многих заболеваний лежат нарушения функционирования ферментов в клетке — энзимопатии. Различают первичные (наследственные) и вторичные (приобретённые) энзимопатии. При первичных энзимопатиях дефектные ферменты наследуются, в основном, по аутосомнорецессивному типу. Гетерозиготы, чаще всего, не имеют фенотипических отклонений. Первичные энзимопатии обычно относят к метаболическим болезням, так как происходит нарушение определённых метаболических путей.
Фенилкетонурия (ФКУ). Фенилаланин (ФА) принадлежит к числу незаменимых аминокислот. Только часть ФА используется для синтеза белков; основное количество этой аминокислоты окисляется до тирозина. Реакцию гидроксилирования ФА катализирует фермент – фенилаланингидроксилаза. Причина ФКУ – недостаточность этого фермента. В результате этого нарушения ФА не превращается в тирозин, из которого образуется фенилпировиноградной кислоты (ФПВК), которая выделяется с мочой и потом, вследствие чего от больных детей исходит «мышиный» запах. Высокая концентрация ФПВК приводит к нарушению формирования миелиновой оболочки вокруг аксонов в ЦНС. ФПВК является нейротропным ядом, в результате чего повышаются возбудимость, тонус мышц, развиваются гиперрефлексия, тремор, судорожные эпилептиформные припадки. Позже присоединяются нарушения высшей нервной деятельности, умственная отсталость, микроцефалия. У больных детей наблюдается слабая пигментация из-за нарушения синтеза меланина. Диагностика заболевания осуществляется биохимическими методами: ещё до развития клинической картины в моче определяется ФПВК, в крови — высокое содержание фенилаланина. В родильных домах обязательно проводится скрининг-тест на фенилкетонурию.
Алкаптонурия. Нарушено окисление гомогентизиновой кислоты в тканях (гомогентизиновая кислота — промежуточный метаболит катаболизма тирозина). У таких больных наблюдают недостаточность фермента окисления гомогентизиновой кислоты — диоксигеназы гомогентизиновой кислоты, приводящей к развитию заболевания. В результате увеличиваются концентрация гомогентизиновой кислоты и выведение её с мочой. В присутствии кислорода гомогентизиновая кислота превращается в соединение чёрного цвета — алкаптон. Поэтому моча таких больных на воздухе окрашивается в чёрный цвет. Алкаптон также образуется и в биологических жидкостях, оседая в тканях, коже, сухожилиях, суставах. При значительных отложениях алкаптона в суставах нарушается их подвижность.
Альбинизм. При альбинизме нарушен синтез в меланоцитах пигментов — меланинов. Меланин находится в коже, волосах, радужке, пигментном эпителии сетчатки глаза и влияет на их окраску. При альбинизме наблюдают слабую пигментацию кожи, светлые волосы, красноватый цвет радужки глаза из-за просвечивающих капилляров. Проявление альбинизма связано с недостаточностью фермента тирозингидроксилазы (тирозиназы) — одного из ферментов, катализирующего метаболический путь образования меланинов.
Болезнь Хартнупа. Причина — недостаточность белков-переносчиков триптофана в кишечной стенке и почечных канальцах. Наблюдаются:повышенное образование индикана, который превращается в индиго синего цвета – симптом «голубых пеленок»; аминоацидурия; признаки пеллагры. нарушение психики. Лечение: полноценная белковая диета, прием никотинамида, предохранение от УФО.
ТВОРЧЕСКАЯ РАБОТА СТУДЕНТОВ. Тема реферативного сообщения/презентации —«Энзимопатии обмена аминокислот. Генетический скрининг новорожденных».
Лекция № 3. Конечные продукты обмена простых белков.
План лекции:
1. Реакции образования аммиака в организме. Токсичность аммиака.
2. Способы обезвреживание аммиака в организме. Орнитиновый цикл. Диагностическое значение определения мочевины.
3. Обмен креатина. Синтез креатина, образование креатинфосфата и креатинина. Диагностическое значение определения креатина, креатинина и креатинкиназы.
Содержание лекционного материала:
1. Реакции образования аммиака в организме. Токсичность аммиака.
Источники аммиака в организме:
a) Дезаминирование аминокислот.
b) Дезаминирование биогенных аминов.
c) Дезаминирование пуриновых и пиримидиновых азотистых оснований.
d) Гниение аминокислот в кишечнике.
Катаболизм аминокислот в тканях происходит постоянно со скоростью ∼100 г/сут. При этом в результате дезаминирования аминокислот освобождается большое количество аммиака. Значительно меньшие количества его образуются при дезаминировании биогенных аминов и нуклеотидов.
Часть аммиака образуется в кишечникев результате действия бактерий на пищевые белки (гниение белков вкишечнике) и поступает в кровь воротной вены. Концентрация аммиака в крови воротной вены существенно больше, чем в общем кровотоке. В печени задерживается большое количество аммиака, что поддерживает низкое содержание его в крови. Концентрация аммиака в крови в норме редко превышает 25-40 мкмоль/л.
Аммиак — токсичное соединение. Даже небольшое повышение его концентрации оказывает неблагоприятное действие на организм, и прежде всего на ЦНС. Так, повышение концентрации аммиака в мозге до 0,6 ммоль вызывает судороги. К симптомам гипераммониемии относят тремор, нечленораздельную речь, тошноту, рвоту, головокружение, судорожные припадки, потерю сознания. В тяжёлых случаях развивается кома с летальным исходом.
2. Способы обезвреживание аммиака в организме.
2.1. Локальный – в тканях. Основной реакцией связывания аммиака, протекающей во всех тканях организма, является синтез глутамина под действием глутаминсинтетазы:
Глутамин легко транспортируется через клеточные мембраны путём облегчённой диффузии (для глутамата возможен только активный транспорт) и поступает из тканей в кровь. Основными тканями-поставщиками глутамина служат мышцы, мозг и печень. С током крови глутамин транспортируется в почки и печень.
В мозге и некоторых других органах может протекать восстановительное аминирование α-кетоглутаратапод действием глутаматдегидрогеназы, катализирующей обратимую реакцию. Однако этот путь обезвреживания аммиака в тканях используется слабо, так как глутаматдегидрогеназа катализирует преимущественно реакцию дезаминирования глутамата.
2.2. 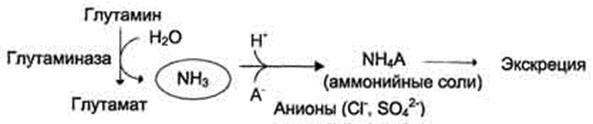 Образование аммонийных солей. В почкахпроисходит гидролиз глутамина под действием глутаминазы с образованием аммиака. Образующийся аммиак нейтрализует кислые продукты обмена и в виде аммонийных солей, которые экскретируются с мочой:
Образование аммонийных солей. В почкахпроисходит гидролиз глутамина под действием глутаминазы с образованием аммиака. Образующийся аммиак нейтрализует кислые продукты обмена и в виде аммонийных солей, которые экскретируются с мочой:
2.3. Орнитиновый цикл. Диагностическое значение определения мочевины.
Мочевина — основной конечный продукт азотистого обмена,в составе которого из организма выделяется до 90% всего выводимого азота. В норме концентрация мочевины в сыворотке/плазме крови – 2,61-8,35 ммоль/л (уреазный метод).
Мочевина выводится из организма с мочой. Азот мочевины составляет 85-90% от общего азота мочи. В норме у взрослых выводится мочевины с мочой 20-35 г/сут (330-580 ммоль/сут).
При повышении количества потребляемых с пищей белков экскреция мочевины увеличивается. Мочевина синтезируется только в печени. В 40-х годах XX века немецкие биохимики Г. Кребс и К. Гензелейт установили, что синтез мочевины представляет собой циклический процесс, состоящий из нескольких стадий, ключевым соединением которого, замыкающим цикл, является орнитин. Поэтому процесс синтеза мочевины получил название «орнитиновый цикл»,или «цикл Кребса-Гензелейта».
Суммарное уравнение синтеза мочевины:
NH3 + Аспартат + СО2 + 3 АТФ + 2 Н2О → Мочевина + Фумарат + 2 АДФ +
В первой реакции процесса аммиак связывается с СО2 с образованием карбамоилфосфата, при этом затрачиваются 2 молекулы АТФ. Реакция происходит в митохондриях гепатоцитов под действием фермента карбамоилфосфатсинтетазы I. Карбамоилфосфат затем включается в орнитиновый цикл и используется для синтеза мочевины.
Биологическая роль орнитинового цикла
Орнитиновый цикл в печени выполняет 2 функции:
· превращение азота аминокислот в мочевину, которая экскретируется и предотвращает накопление токсичных продуктов, главным образом аммиака;
· синтез аргинина и пополнение его фонда в организме.
Нарушение реакций обезвреживания аммиака может вызвать повышение содержания аммиака в крови— гипераммониемию, что оказывает токсическое действие на организм. Причинами гипераммониемии могут выступать как генетический дефект ферментов орнитинового цикла в печени, так и вторичное поражение печени в результате цирроза, гепатита и других заболеваний.
Все нарушения орнитинового цикла приводят к значительному повышению в крови концентрации аммиака, глутамина и аланина.
Гипераммониемиясопровождается появлением следующих симптомов:
- тошнота, повторяющаяся рвота;
- головокружение, судороги;
- потеря сознания, отёк мозга (в тяжёлых случаях);
- отставание умственного развития (при хронической врождённой форме).
Клинико-диагностическое значение исследования концентрации мочевины в сыворотке крови и моче.
Определение концентрации мочевины в сыворотке крови используется для оценки функции печени и выделительной функции почек. Критическая концентрация мочевины в крови: ˃28,0 ммоль/л. Причины увеличения концентрации мочевины в сыворотке крови:острые или хронические заболевания почек; обтурация мочевыводящих путей; снижение тока крови через почечные клубочки (застойная сердечная недостаточность, обширные ожоги, тяжелая диарея и рвота, инфаркт миокарда и др.); повышенный катаболизм белков (длительное голодание, кровотечения в ЖКТ, инфекции, канцерогенез, сахарный диабет и др.); снижение концентрации Cl — . Причины снижения концентрации мочевины в сыворотке крови: функциональная недостаточность печени (гепатит, цирроз, отравления мышьяком и др.); энзимопатии синтеза мочевины; повышенная скорость клубочковой фильтрации, обусловленная беременностью, чрезмерным объемом внутривенных инфузий, неадекватной секрецией антидиуретического гормона; мальабсорбция (целиакия). Влияющие факторы. Увеличение концентрации: диета с высоким содержанием белка, прием нефротоксичных лекарств, повышенный распад тканей при гипертиреозе, приеме глюкокортикоидов. Уменьшение концентрации:вегетарианская диета.
Определение концентрации мочевины в моче позволяет контролировать азотистый баланс у больных в тяжелом состоянии, получающих зондовое энтеральное и парентеральное питание. Корректно оценить азотистый баланс с использованием этого теста можно лишь при отсутствии тяжелой печеночной или почечной патологии. Увеличением концентрации мочевины в моче, указывающим на отрицательный азотистый баланс, сопровождаются: послеоперационный период, лихорадка; гипертиреоз; тяжелые острые или хронические соматические заболевания. Уменьшением, указывающим на положительный азотистый баланс, сопровождаются: беременность; период роста у детей; период восстановления после тяжелых заболеваний.
3. Обмен креатина. Синтез креатина, образование креатинфосфата и креатинина. Диагностическое значение определения креатина, креатинина и креатинкиназы.
Синтез креатина. Креатин необходим для образования в мышцах высокоэнергетического соединения — креатинфосфата. Синтез креатина идёт в 2 стадии с участием 3-х аминокислот: аргинина, глицина и метионина. В почкахобразуется гуанидинацетат при действии глицинамидинотрансферазы.Затем гуанидинацетат транспортируется в печень,где происходит реакция его метилирования:
SAM – S-аденозилметионин – активная форма метионина – донор СН3-группы.
 Креатин с кровотоком переносится в мышцы и клетки мозга,где из него образуется высокоэнергетическое соединение — креатинфосфат.
Креатин с кровотоком переносится в мышцы и клетки мозга,где из него образуется высокоэнергетическое соединение — креатинфосфат.
 |
Эта реакция легко обратима и катализируется ферментом креатинкиназой. Обнаружено три изоферментные формы креатинкиназы (КК-ВВ – мозговой тип, КК-МВ – сердечный тип, КК-ММ – мышечный тип). Измерение активности общей креатинкиназы и ее изоферментов используется в лабораторной диагностике.
Креатинфосфат играет важную роль в обеспечении энергией работающей мышцы в начальный период.
Креатинин– это конечный продукт неферментативного превращения креатина и креатинфосфата, участвующих в энергообеспечении мышечного сокращения:
Креатинин выводится из организма через почки с мочой, относится к беспороговым веществам, которые поступают в мочу путем фильтрации и не реабсорбируются в канальцах. Нормы креатинина в сыворотке крови у взрослых: мужчины – 40-100 мкмоль/л, женщины – 44-88 мкмоль/л; креатинина в моче у взрослых — 4,4-17,7 ммоль/сут.
Клинико-диагностическое значение исследования концентрации креатина и креатинина в сыворотке крови и моче. Гиперкреатининемия – увеличение уровня креатинина в крови обусловлено как усиленным образованием, так и задержкой его в организме. Критическая концентрация креатинина в сыворотке крови: ˃400 мкмоль/л. Ретенционная гиперкреатининемия обусловлена нарушением (острым и хроническим) функции почек любого происхождения. Продукционная гиперкреатининемия отмечается при кишечной непроходимости, декомпенсации деятельности ССС, пневмонии, лихорадочных состояниях, гипертиреозе, голодании, усиленной мышечной работе. Снижение уровня креатинина в сыворотке крови коррелирует с уменьшением мышечной массы (мышечные дистрофии и атрофии, параличи и др.). Увеличение суточного выведения креатинина с мочойуказывает на увеличение почечной фильтрации и/или повышенное образование креатинина в скелетных мышцах. Увеличением концентрации сопровождаются: усиленная мышечная работа, лихорадочные состояния, пневмония, акромегалия, сахарный диабет, острые инфекционные заболевания. Уменьшение выведения креатинина с мочой наблюдается при мышечной атрофии, параличах, хронических заболеваниях почек, острой почечной недостаточности, голодании. В моче помимо эндогенного креатинина содержится экзогенный креатинин, поступающий в организм с мясной пищей. На концентрацию креатинина в крови и моче влияет характер диеты: преимущественно мясная диета приводит к увеличению, и наоборот, вегетарианская диета – к снижению концентрации креатинина в крови и моче.
Креатинемия (креатин + греч. haima кровь) — наличие в крови креатина. В плазме взрослого человека концентрации креатина в норме составляет 15,25—76,25 мкмоль/л. Повышение уровня креатина в крови (гиперкреатинемия) может быть при избыточном употреблении мяса, а также при заболеваниях мышечной системы, при кишечной непроходимости. поражениях печени. Креатинв моче взрослых людей в норме практически отсутствует. В первые годы жизни ребенка возможна «физиологическая креатинурия». По-видимому, появление креатина в моче детей раннего возраста обусловлено усиленным синтезом креатина, опережающим развитие мускулатуры. Некоторые исследователи к физиологическим явлениям относят и креатинурию стариков, которая возникает как следствие атрофии мышц и неполного использования образующегося в печени креатина. При концентрации креатина в плазме крови свыше 122,0 мкмоль/л он начинает выделяться с мочой. Наибольшее содержание креатина в моче наблюдается при патологических состояниях мышечной системы и прежде всего при миопатии, или прогрессирующей мышечной дистрофии. Принято считать, что креатин в моче (креатинурия) больных миопатией может появляться в результате нарушения в скелетной мускулатуре процессов его фиксации (удержания) и фосфорилирования. Если нарушен процесс синтеза фосфокреатина, то не образуется и креатинин; содержание последнего в моче резко снижается. В результате креатинурии и нарушения синтеза креатинина резко повышается креатиновый показатель мочи: (количество креатина + количество креатинина)/(количество креатинина). В норме этот показатель близок к 1,1.
ТВОРЧЕСКАЯ РАБОТА СТУДЕНТОВ. Тема реферативного сообщения презентации«Энзимопатии цикла синтеза мочевины. Лабораторная диагностика гипераммониемии».
Дата добавления: 2015-10-01 ; просмотров: 1545 | Нарушение авторских прав
источник
Альбинизм – это наследственная болезнь, которая очень ярко проявляется в белом «неестественном» цвете кожи, волос и глаз. Если среди азиатов и европейцев данная патология – редкое явление, по статистике 1 человек на 20 000 только может страдать этим заболевание, то в Африке, вопреки всем законам логики, альбиносом является 1 человек из 4000 в Танзании и ЮАР, а в Нигерии -1 из 1100.
Африканцы до сих пор живут во тьме суеверий и невежества относительно этой болезни. С одной стороны они считают альбинизм – проклятием, с другой стороны, что он приносит удачу. Самое страшное то, что в Танзании до сих пор идет торговля органами альбиносов, части их тела продают шаманам и колдунам, которые делают различные снадобья из органов и «рекламируют» их от всяких бед. С целью спасения людей альбиносов создаются закрытые интернаты, за стены которых они не могут и не хотят выходить в целях своей же безопасности.
Настолько ли страшна эта болезнь? Откуда появляется это светлый ген у негров? Обо всем этом мы будем разбираться в нашей статье.
Известно, что в данной болезни задействован механизм мутации генов, поэтому в этом нам поможет разобраться 2 науки генетика и биохимия.
В нашем организме присутствует аминокислота тирозин, который отвечает за синтез пигмента меланина. Синтез меланина включает в себя следующие стадии:
- Тирозин «доставляют» через мембрану меланосом к ферментам, которые будут взаимодействовать с аминокислотой. «Доставку» осуществляет Р-белок.
- Тирозин вступает в обменные реакции с ферментом тирозиназа, который отвечает за количество вырабатываемого пигмента;
- Далее идет взаимодействие ферментами TRP 2 и TRP1, которые образовывают эумеланин и феомеланин. За черный и коричневый цвет кожи несет ответственность эумелани, а за желтый и красный – феомеланин.
Биохимическая причина альбинизма заключается в ферменте тирозиназа. Этот фермент провоцирует развитие тирозиназа- позитивного/негативного альбинизма. Негативный развивается, если в волосяных луковицах отсутствует тирозиназа, в противном случае развивается позитивный.
Генетика определяет цвет кожи человека, до конца неизвестно, какие именно мутации происходят с генами, отвечающими за выработку тирозиназы, потому что существует альбинизм с нормальным синтезом этого фермента.
Ученые считают, что механизм развития альбинизма зависит от разнообразных генных мутаций, связанных также с ответственными генами за фермент энзим и других.
Ребенок рождается данной патологией, в случае если оба родителя имеют дефектный ген, т.е наследование осуществляется по аутосомно-рецессивному типу.
Результат этих мутаций один, в коже отсутствует пигмент меланин, который защищает кожу и глаза от ультрафиолетового излучения.
Существует различные виды альбинизма, которые определяют степень последствия для каждого больного человека.
В зависимости от вовлеченности клеток в патологический процесс выделяют глазной и 3 типа глазокожного альбинизма.
Глазо-кожный альбинизм первого типа (ГКА 1) возникает из-за того, что 11 хромосоме в результате мутации происходит трансформация здорового гена в кодирующий ген, который препятствует полному либо частичному синтезу фермента тирозиназы. Если происходит полное прекращение синтеза, то это относится к подтипу 1 А, который имеет самые тяжелые последствия и проявления. При данном типе ребенок рождается с абсолютно бело-молочной кожей и волосами, на теле ребенка нет ни одной родинки, глаза голубые, его кожа никогда не сможет загореть.
Подтип 1В характеризуется лишь частичным синтезом тирозиназы, она вырабатывается лишь в количестве 20-30% от нормы. Симптомы присутствуют с самого рождения, но наблюдается малая пигментация кожи, ребенок может иметь желтоватого цвета волоса, так называемый желтый или платиновый альбинизм. С годами кожа и волосы могут приобретать цвет, в ряде случаев кожа получает небольшой загар и могут появляться родинки.
Отсутствие меланина в пигментом слое сетчатки глаза приводит к тому, что развивается в обоих подтипах нистагм – быстрое неконтролируемое движение глаз, острота зрения не превышает 0,1-0,2 при норме 1, появляется светобоязнь.
У подтипа 1В существует свой подтип температуро-чувствительный (теплозависимый) альбинизм. Данная форма формируется под влиянием температуро-чувствительного фермента, который образовался в результате мутации гена тирозиназы. В отличие от здорового фермента, теплозависимая тирозиназа «капризничает» при повышении температуры. Проявляется это в том, что в холодных местах тела (конечности) волосы темные, т.е фермент участвует в синтезе меланина, в теплых же местах – под мышками, на голове – они остаются белыми (меланин не вырабатывается), со временем они могут пожелтеть.
Когда ребенок рождается, температура его тела более высокая, чем температура взрослых людей, поэтому теплозависимая форма альбинизма проявляется со временем. Пигмент начнет вырабатываться на конечностях в случае «похолодания», но изменения будут происходить только на коже, глаза являются «теплолюбивым» органом.
Глазо-кожный альбинизм второго типа (ГКА 2) не затрагивает ген, контролирующий синтез тирозиназы, т.е. фермент вырабатывается в достаточном количестве, он находится в волосяных луковицах. Мутация происходит в гене, находящимся на хромосоме 25 и контролирующем мембранный Р –белок (он отвечает за транспортировку фермента и ионов).
В всех странах этот тип наиболее распространен. Отличительными признаками этой разновидности является то, что с возрастом кожа, волосы начинают темнеть, т.е. происходит пигментация, количество вырабатываемого пигмента зависит от расовой принадлежности, по мере увеличения пигмента повышается острота зрения.
Для европейцев характерно появление веснушек, родинок, пигментных пятен. У негроидной расы возможно появление коричневой окраски кожи и волос, на чаще цвет волос у них желтый.
Характерным является развитие амблиопии — обратимого нарушения глаз, при котором функционирует только один глаз.
Глазо-кожный альбинизм третьего типа (ГКА 3) – это достаточно редкий вариант альбинизма, связанный с мутацией гена, контролирующего выработку TRP1 фермента, в результате снижается концентрация этого фермента и происходит купирование синтеза эумеланина, отвечающего за коричневую окраску. Проявления этой болезни схожи с проявлениями первого типа, но они более умеренные. У новорожденного присусвует слабая окраска кожи и волос, с возрастом может произойти увеличение в коже пигмента. Кожа воспринимает загар и на ней присутствуют родинки, глаза страдают горазда меньше, чем в вышеописанных типах.
Глазной альбинизм, как следует из названия, от него страдают только глаза, кожа может быть чуть светлее, чем у родителей. Отсутствие пигмента в сетчатке приводит к тому, что ухудшается зрение, развиваются рефракционные дефекты, нистагм , косоглазие и много других зрительных аномалий.
Отличие глазного альбинизма от глазо–кожных типов в том, что носителями деформированного гена являются женщины, а болеют мужчины. Женщины лишь могут иметь слабые проявления болезни, в виде пятен на глазном дне и прозрачной радужки.
Мутация происходит с геном, отвечающим за формирование гликобелка, в результате мутации он кодируется и меланосомы, которые содержат меланин не созревают.
Кроме того крайне редко встречаются формы альбинизма, совмещенные с различными патологиями организма, выделяют следующие синдромы:
Синдром Хермански—Пудлака — заболевание, характеризуется альбинизмом и низкой свёртываемостью крови. Цвет кожи проявляется индивидуально от почти нормального до белого, присутствуют различные зрительные дефекты. Оба родителя имеют дефектный ген.
Синдром Чедиака—Хигаси — заболевание, которое характеризуется частичным альбинизмом и низким иммунитетом.
Мутации, происходящие в генах, отвечающих за производство Р-белка и вызывающих ГКА 2 типа, могут быть частью мутации происходящей в генах PWS и AS, а эти мутация вызывают Синдром Прадера-Вилли и синдром Ангельмана, около 1 % людей с этими синдромами имеют также альбинизм.
Для этих синдромов характерны умственные нарушения, пониженное давление и др.
По степени вовлеченности кожных покровов различают:
- Полный альбинизм – это весь кожный покров страдает от болезни;
- Частичны, который имеет название пьебалдизм, характерными признаками у новорожденного является белая(седая) прядь волос и белые пятна различного размера по всему телу, в некоторых пятнах могут быть вкрапления нормальной кожи. Эти пятна остаются с человеком на всю жизнь, солнце их может чуть-чуть подкорректировать. Отличие от витилиго заключается в том, что это врожденная болезнь, а не приобретенная. В некоторых случаях частичный альбинизм сочетается с глухотой и неврологическими отклонениями.
Эта патология сама по себе не угрожает жизни пациента, но солнце несет прямую угрозу для человека-альбиноса, поэтому основная задача защитит человека от солнца, потому что иначе больной долго не проживет. В Африканских бедных странах длительность их жизни составляет около 40 лет из-за того, что просто нет солнцезащитных кремов. Врачи, обследовавшие африканцев-альбиносов, заметили, что к 16-18 годам они практически все слепые и у многих развивается рак кожи.
Основное лечение включает в себя – солнцезащитный крем, солнцезащитные очки, полный запрет на загар.
Не смотря на то, что биохимия и генетика возникновения альбинизма изучается специалистами, на данный момент исправить мутацию невозможно, поэтому эффективного лечения этой болезни нет.
источник
Тирозин, помимо участия в синтезе белков, является предшественником гормонов надпочечников адреналина, норадреналина, медиатора дофамина, гормонов щитовидной железы тироксина и трийодтиронина, пигментов. Нарушения катаболизма тирозина многочисленны и называются тирозинемии.
Тирозинемия типа I (гепаторенальная тирозинемия) возникает при недостаточности фумарилацетоацетат-гидролазы . При этом накапливается фумарилацетоацетат и его метаболиты (сукцинилацетон), поражающие печень и почки. Частота заболевания 1:100-120 тыс новорожденных.
Существует две формы – острая и хроническая.
Острая форма составляет большинство случаев этой тирозинемии с началом в возрасте 2-7 мес и смертью 90% больных в возрасте 1-2 года из-за недостаточности печени.
К симптомам относится гипотрофия, рвота, «капустный запах» от тела и мочи, задержка развития, кровоточивость, диарея, мелена, гематурия, желтуха, анемия, периферические невропатии и параличи, кардиомиопатия, слабость мышц, дыхательные нарушения. Отмечают гипогликемию вследствие гиперплазии островковых клеток поджелудочной железы.
При хронической форме болезнь развивается позднее, медленнее прогрессирует. Продолжительность жизни около 10 лет.
Наблюдаются гипотрофия, узелковый цирроз печени и печеночная недостаточность, множественные дефекты почечной канальцевой реабсорбции с появлением синдрома Фанкони (щелочная рН мочи, глюкозурия, протеинурия), аминоацидурия, лейкопения, тромбоцитопения.
Из-за поражения печени и почек возникают проявления рахитоподобных заболеваний (остеопороз, остеомаляция). В результате печеночной недостаточности возникают симптомы, напоминающие острую порфирию. Непостоянными признаками являются умственная отсталость и неврологические изменения.
Лекарственным средством является нитизинон , конкурентный ингибитор 4-гидроксифенилпируват-диоксигеназы. В результате прекращается образование гомогентизиновой кислоты и ее дальнейший распад. Также используется диета со снижением количества фенилаланина и тирозина, инъекции глутатиона.
Гораздо более редкое заболевание по сравнению с тирозинемией I типа.
Тирозинемия типа II (глазокожная тирозинемия) возникает при недостаточности тирозин-аминотрансферазы .
Наблюдается задержка умственного и физического развития, микроцефалия, катаракты и кератоз роговицы (псевдогерпетический кератит), гиперкератоз кожи, членовредительство, нарушение тонкой координации движений.
Поражения почек и печени не наблюдается.
Эффективна диета с низким содержанием тирозина, при этом поражения кожи и роговицы быстро исчезают.
Тирозинемия III типа – результат генетического дефекта 4-гидроксифенилпируват-диоксигеназы . Зафиксировано лишь несколько случаев этой болезни.
Характерные особенности включают умеренную умственную отсталость, судороги и периодическую потерю равновесия и координации (прерывистая атаксия).
Тирозинемия новорожденных – результат кратковременного снижения активности 4- гидроксифенилпируват-диоксигеназы . Чаще наблюдается у недоношенных детей.
Наблюдается сниженная активность и летаргия. Аномалия считается безвредной. Дефицит аскорбиновой кислоты усиливает клиническую картину.
Диета со снижением количества белка, фенилаланина, тирозина и высокие дозы аскорбиновой кислоты (100 мг/день).
Генетическая аутосомно-рецессивная энзимопатия. В основе заболевания лежит снижение активности печеночного фермента гомогентизат-оксидазы , в результате в организме накапливается гомогентизиновая кислота.
Так как гомогентизат на воздухе окисляется и полимеризуется в меланиноподобное соединение, то наиболее частым и постоянным симптомом является темная моча, на пеленке и нижнем белье остаются темно-коричневые пятна. Другим образом в детском возрасте болезнь не проявляется.
С возрастом гомогентизиновая кислота, накапливается в соединительно-тканных образованиях, склерах и коже, вызывает шиферно-глубокий оттенок ушного и носового хрящей (охроноз), окрашивает одежду, контактирующую с потеющими участками тела (подмышки).
Из-за связывания гомогентизата с коллагеном ухудшается состояние соединительной ткани, что делает хрупкими хрящевые образования. После 30 лет развивается дегенеративный артрит позвоночника и крупных суставов (бедренные, коленные), межпозвонковые пространства сужены, снижается минеральная плотность костей. Может наблюдаться поражение почек и сердца.
Хотя эффективные способы неизвестны, по аналогии с другими аминокислотными нарушениями рекомендуется с раннего возраста ограничить потребление фенилаланина и тирозина, что должно препятствовать развитию охроноза и суставных нарушений. Назначают большие дозы аскорбиновой кислоты для снижения связывания гомогентизиновой кислоты в соединительной ткани. Предлагается использовать препарат нитизинон , конкурентный ингибитор 4-гидроксифенилпируват-диоксигеназы.
Заболевание обусловлено полным или частичным дефектом синтеза фермента тирозиназы (частота 1:20000), необходимой для синтеза диоксифенилаланина и далее меланинов в пигментных клетках.
При полном отсутствии фермента наблюдается тотальная депигментация кожи, волос, глаз, причем окраска одинакова для всех расовых групп и не меняется с возрастом. Кожа не загорает, совершенно отсутствуют невусы, какие-либо пигментные пятна, развиваются фотодерматиты. Сильно выражены нистагм, светобоязнь, дневная слепота (т.к. имеется депигментация сетчатки и ускоренный распад родопсина), красный зрачковый рефлекс.
При частичной недостаточности фермента отмечаются светло-желтые волосы, слабо пигментированные родинки, очень светлая кожа.
Рекомендуется использовать различные средства защиты от ультрафиолетовых лучей.
Биохимической причиной паркинсонизма (частота после 60 лет 1:200) является низкая активность тирозин-гидроксилазы или ДОФА-декарбоксилазы в нервной ткани, при этом развивается дефицит нейромедиатора дофамина и накопление тирамина.
Наиболее распространенными симптомами являются ригидность мышц, скованность движений, тремор и самопроизвольные движения.
Требуется систематическое введение лекарственных аналогов дофамина и применение ингибиторов моноаминоксидазы.
источник
Альбинизм – группа наследственных патологий, характеризующихся нарушениями или полным отсутствием пигментации кожи, волос, радужной оболочки глаза. Основными симптомами заболевания является очень светлая кожа и волосы, голубой или красноватый цвет глаз, в ряде случаев могут быть нарушения зрения. Диагностика альбинизма производится на основании настоящего статуса пациента, а также генетических исследований. Специфического лечения альбинизма на сегодняшний день нет, используют паллиативную терапию (коррекция зрения), а также существует ряд рекомендаций больным, как вести себя на солнце, защищать кожу и снизить вероятность осложнений.
Альбинизм – совокупность генетических патологий, при которых нарушаются процессы формирования или накопления пигмента меланина в клетках кожи, ее придатков, радужной оболочке и сетчатке глаза. Это состояние известно с древних временен, поражает лиц любой национальности или расы. Однако частота встречаемости альбинизма отличается у разных народностей – она составляет от 1:10000 до 1:2000000. Также этот показатель неодинаков для разных форм заболевания, которые в последние годы классифицируются по генетическим признакам. Ранее выделяли всего две формы альбинизма (глазную и кожно-глазную), хотя современная генетика различает, по меньшей мере, семь различных типов патологии. Кроме того, существует ряд наследственных заболеваний, симптомокомплекс которых, помимо всего прочего, включает в себя и альбинизм – например, синдром Чедиака-Хигаси, болезнь Германского-Пудлака.
Основная причина развития альбинизма – нарушения метаболизма аминокислоты тирозина, и, как следствие, полный блок или ослабление синтеза и отложения пигмента меланина. К этому состоянию могут привести различные мутации генов, которые прямо или косвенно участвуют в процессе образования меланина. Например, наиболее тяжелая форма альбинизма – глазокожная 1А – обусловлена сложной мутацией гена alb-OCA1, расположенного на 11-й хромосоме. Он кодирует последовательность фермента тирозиназы, и при nonsense-мутациях его производство в организме полностью останавливается. В результате образование меланина также полностью прекращается, что и становится причиной тяжелого глазокожного альбинизма. Наследуется это состояние по аутосомно-рецессивному механизму.
Другой тип этого заболевания – глазокожный альбинизм 1В — обусловлен нарушением того же гена alb-OCA1, однако при этом его функционирование продолжается. С данной патологией ассоциировано более 50-ти мутаций вышеуказанного гена, каждая из которых в разной мере влияет на активность тирозиназы. Поэтому выраженность симптомов при глазокожном альбинизме 1В также очень вариабельна – от почти полного отсутствия меланина в тканях до слегка более светлого оттенка кожи и волос. В ряде случаев больные с такой формой патологии способны загорать, у них с возрастом темнеют волосы, могут появляться пигментные невусы. Интересным подтипом данного заболевания является температурно-чувствительный альбинизм, при котором активность тирозиназы резко падает при температуре свыше 37 градусов. Это приводит к тому, что пигментация сильнее проявляться на более холодных участках тела – кисти, стопы. Тогда как голова, глаза, подмышечные впадины часто остаются практически без пигмента.
Глазокожный альбинизм типа 2 является наиболее распространенной разновидностью данной патологии. Однако при этом генетические нарушения не затрагивают синтез тирозиназы, который сохраняется на достаточном уровне, активность и структура фермента также не страдают. Альбинизм такого типа обусловлен мутацией гена, расположенного на 15-й хромосоме. Предположительно, он кодирует протеин (Р-белок) мембраны меланосом, который отвечает за транспорт тирозина. При этой форме альбинизма проявления дефицита меланина также очень вариабельны, к тому же, пигментация может усиливаться со временем. Причины такого явления до сих пор не выяснены. Наследуется глазокожный альбинизм типа 2 по аутосомно-рецессивному типу.
Другая форма заболевания, глазокожный альбинизм типа 3 встречается почти исключительно у негроидной расы. При нем генетическими исследованиями выявлены мутации гена TRP-1, расположенного на 9-й хромосоме. Аналогичный ген у мышей отвечает за коричневую окраску шерсти, его функции у человека достоверно неизвестны. Предполагается, что он контролирует образование черной фракции меланина (эумеланина), а нарушение его структуры ведет к преимущественному синтезу коричневой разновидности пигмента. Как и другие глазокожные формы альбинизма, тип 3 передается по аутосомно-рецессивному механизму.
Каждый тип альбинизма характеризуются не только исчезновением меланина из кожи и ее придатков, но и зрительного аппарата глаза – радужной оболочки и пигментного слоя. Это приводит к нарушениям рефракции и прозрачности роговицы, астигматизму и косоглазию, фовеолярной гипоплазии сетчатки. Имеются формы альбинизма (так называемые глазные типы), которые характеризуются только поражением органов зрения. Наиболее распространенная форма глазного альбинизма передается по рецессивному типу и сцеплена с Х-хромосомой. Она обусловлена мутацией гена GPR143, кодирующего рецептор к G-белку меланоцитов глаз. В результате этого нарушаются процессы формирования меланосом, что и становится причиной развития глазного альбинизма. В 1970-м году была также выявлена аутосомно-рецессивная форма этого заболевания, однако патогенез данного типа до сих пор не определен – часть (14%) таких больных имели мутации гена alb-OCA1, другие (36%) – нарушения в гене Р-белка. Почти у половины пациентов с аутосомно-рецессивным глазным альбинизмом выявить генетическую причину заболевания не удалось.
Ранее все случаи альбинизма разделяли только по фенотипическим проявлениям – полный и неполный. К первому относились все типы глазокожного альбинизма, которые характеризуются выраженными нарушениями пигментации глаз, кожи и ее придатков. К неполным формам относили глазные типы заболевания, а также те разновидности патологии, которые приводили к пятнистости кожи. В настоящее время чаще используют генетическую классификацию, в рамках которой выделяют такие виды альбинизма:
- Глазокожный альбинизм тип 1А – его причиной выступает nonsense-мутация гена alb-OCA1, которая попросту «выключает» его экспрессию. В результате этого полностью останавливается синтез тирозиназы в организме.
- Глазокожный альбинизм тип 1В – так же, как и в предыдущем случае, он обусловлен мутациями гена alb-OCA1, однако при этом его экспрессия возможна. В результате синтезируется дефектный фермент тирозиназа с разной степенью активности. Выраженность проявлений такого альбинизма зависит от типа мутации гена.
- Температурно-чувствительный глазокожный альбинизм – является разновидностью типа 1В, характеризуется изменчивой активностью тирозиназы, которая зависит от температуры. Кожные проявления умеренные, тогда как офтальмологические нарушения могут быть значительно выражены. Данные особенности такого альбинизма обусловлены более высокой температурой глаз – следовательно, тирозиназа в них менее активна.
- Глазокожный альбинизм тип 2 – вызван мутацией гена, кодирующего Р-белок, являющийся элементом мембраны внутриклеточных меланосом. В результате транспорт тирозина в клетке нарушается, и синтез меланина не происходит даже при нормальной активности тирозиназы.
- Глазокожный альбинизм тип 3 – является следствием мутаций гена TRP-1, который, предположительно, контролирует образование эумеланина. Встречается только у африканцев, вызывает развитие коричневой окраски кожи и волос и умеренные офтальмологические нарушения.
- Глазной альбинизм рецессивный и связанный с Х-хромосомой. Он обусловлен мутацией гена GPR143, отвечающего за некоторые элементы внутриклеточной передачи информации.
- Аутосомно-рецессивный глазной альбинизм – его пока не удалось связать с конкретными генетическими нарушениями. Предполагается, что часть случаев такого заболевания является глазными формами глазокожных типов патологии – 1В и 2.
Даже в пределах одного генотипа альбинизма возможны значительные различия в степени выраженности симптомов. Это связано с тем, что мутации разного типа неодинаково влияют на выработку меланина.
К главным проявлениям альбинизма относят бледность кожных покровов, особенно заметную при рождении больного ребенка. Нередко кожа имеет розоватый оттенок из-за просвечивающихся кровеносных сосудов, глаза при рождении голубые, но в некоторых ракурсах также могут иметь красноватый цвет. В дальнейшем, в процессе роста, симптомы альбинизма могут несколько изменяться в зависимости от типа заболевания. При типе 1А, который является наиболее тяжелым, синтеза меланина в организме не происходит вообще, поэтому у больного пожизненно сохраняется белый цвет кожи и волос и голубые глаза. Альбинизм типа 1В характеризуется быстрым накоплением в волосах желтого пигмента, поэтому они принимают светло-соломенный цвет, часто с возрастом происходит пигментация ресниц и роговицы глаз.
Температурно-чувствительный альбинизм часто проявляется своеобразным распределением меланина – нормальная пигментация наблюдается на конечностях, тогда как кожа головы остается бледной, волосы также сохраняют белый цвет. Глаза по причине повышенной, нежели на конечностях, температуры остаются у таких больных голубого цвета. Значительной переменчивостью симптомов характеризуется и альбинизм типа 2 – от почти полного отсутствия пигментации до незаметного посветления кожи и волос. Также для такой формы заболевания часто характерно улучшение синтеза меланина с возрастом – начинают темнеть волосы, появляться веснушки, возникать загар. Однако с пребыванием на солнечном свете необходимо быть осторожным – кожа больных альбинизмом крайне чувствительна к ультрафиолетовому облучению, легко возникают ожоги кожи и фотодерматиты.
Характерным симптомом альбинизма является нарушение остроты зрения у больных и другие офтальмологические изменения. Снижение зрения тем выраженнее, чем слабее синтезируется в организме меланин, особенно в роговице и пигментном слое сетчатки. Кроме этого, частыми спутниками альбинизма являются косоглазие, астигматизм, нистагм, которые возникают сразу при рождении или в первые годы жизни. При глазных формах заболевания подобные симптомы проявляются без нарушения пигментации кожи и волос. Из-за отсутствия защитного слоя меланоцитов у больных альбинизмом часто возникает светобоязнь, иногда переходящая в дневную слепоту.
Определение альбинизма во многих случаях возможно сразу же после рождения больного – дерматолог, оценивая состояние пигментации кожи и волос способен выявить заболевание и приблизительно узнать его разновидность. Дальнейшее наблюдение у этого специалиста необходимо для мониторинга течения патологии и для профилактики возможных осложнений – например, рака кожи. Врач-офтальмолог при альбинизме нередко выявляет прозрачность радужной оболочки, у взрослых больных часто определяется гипоплазия сетчатки в области желтого пятна. Фовеолярный рефлекс резко снижен или отсутствует. У лиц с неполным альбинизмом на глазном дне часто обнаруживаются очаги депигментации. Обнаруживаются и другие нарушения зрения – нистагм, астигматизм, миопия.
Для подтверждения диагноза и уточнения типа патологии врач-генетик может назначить секвенирование ассоциированных с ней генов. Также важную роль играет составление наследственного анамнеза, возможна генетическая диагностика родственников больного для выявления носителей дефектных генов. Редким и дорогостоящим методом диагностики альбинизма является определение тирозиназной активности в тканях (например, в волосяных фолликулах), но это позволяет несколько уточнить прогноз заболевания. Чем лучше сохранена активность этого пигмента в тканях, тем ниже выраженность других симптомов этой патологии. Дифференциальную диагностику альбинизма следует проводить с другими наследственными патологиями, которые сопровождаются сходными кожными и офтальмологическими симптомами. В первую очередь это синдромы Чедиака-Хигаси и Германского-Пудлака, Х-сцепленный ихтиоз, микрофтальмия, болезнь Каллмана.
Специфического лечения альбинизма на сегодняшний день не существует, разработаны лишь профилактические мероприятия, позволяющие улучшить качество жизни больного. Для сохранения существующего уровня зрения необходима защита глаз от солнечного света – это достигается ношением специальных солнцезащитных очков или контактных линз. Появления на ярком солнце необходимо избегать или же защищать кожу специальными кремами и лосьонами. Если придерживаться этих рекомендаций, то в целом прогноз альбинизма благоприятный – больные могут прожить долгую и полноценную жизнь. При этом необходимы регулярные консультации дерматолога и офтальмолога – для профилактики осложнений, таких как рак кожи или отслойка сетчатки.
источник
Причина заболевания — дефект диоксигеназы гомогентизиновой кислоты. Для этой болезни характерно выделение с мочой большого количества гомогентизиновой кислоты, которая, окисляясь кислородом воздуха, образует тёмные пигменты алкаптоны. Это метаболическое нарушение было описано ещё в XVI веке, а само заболевание охарактеризовано в 1859 г. Клиническими проявлениями болезни, кроме потемнения мочи на воздухе, являются пигментация соединительной ткани (охроноз) и артрит. Частота — 2-5 случаев на 1 млн новорождённых. Заболевание наследуется по аутосомнорецессивному типу. Диагностических методов выявления гетерозиготных носителей дефектного гена к настоящему времени не найдено.
Причина метаболического нарушения — врождённый дефект тирозиназы. Этот фермент катализирует превращение тирозина в ДОФА в меланоцитах. В результате дефекта тирозиназы нарушается синтез пигментов меланинов.
Клиническое проявление альбинизма (от лат. albus — белый) — отсутствие пигментации кожи и волос. У больных часто снижена острота зрения, возникает светобоязнь. Длительное пребывание таких больных под открытым солнцем приводит к раку кожи. Частота заболевания 1:20 000.
Нарушение синтеза катехоламинов может вызывать различные нервно-психические заболевания, причём патологические отклонения наблюдаются как при снижении, так и при увеличении их количества.
Заболевание развивается при недостаточности дофамина в чёрной субстанции мозга. Это одно из самых распространённых неврологических заболеваний. При этой патологии снижена активность тирозингидроксилазы, ДОФА-декарбоксилазы. Заболевание сопровождается тремя основными симптомами: акинезия (скованность движений), ригидность (напряжение мышц), тремор (непроизвольное дрожание). Дофамин не проникает через гематоэнцефалический барьер и как лекарственный препарат не используется. Для лечения паркинсонизма предлагаются следующие принципы:
заместительная терапия препаратами-предшественниками дофамина (производными ДОФА) — леводопа, мадопар, наком и др.
подавление инактивации дофамина ингибиторами МАО (депренил, ниаламид, пиразидол и др.).
Депрессивные состояния часто связаны со снижением в нервных клетках содержания дофамина и норадреналина.
Гиперсекреция дофамина в височной доле мозга наблюдается при шизофрении.
Конечные продукты обмена белков:
C, H, O, N , S. – CO2 , H2O, NH3, H2S.
Соли аммония выводятся с мочой:
В почках также происходит гидролиз глутамина под действием глутаминазы с образованием аммиака. Этот процесс является одним из механизмов регуляции кислотно щелочного равновесия в организме и сохранения важнейших катионов для поддержания осмотического давления. Глутаминаза почек значительно индуцируется при ацидозе, образующийся аммиак нейтрализует кислые продукты обмена и в виде аммонийных солей экскретируется с мочой .
Эта реакция защищает организм от излишней потери ионов Na+ и К+, которые также могут использоваться для выведения анионов и утрачиваться. При алкалозе количество глутаминазы в почках снижается.
В почках образуется и выводится около 0,5 г солей аммония в сутки.
В печени – синтез мочевины ( в следующих вопросах будет подробно описано. Честно говоря, странный вопрос – скорее всего надо просто сказать где образуется.)
Вопрос №38. Основные источники и пути обезвреживания аммиака в организме.
Непрямое дезаминирование(основной путь дезаминирования амк)
Окислителное дезаминирование глутамата
Окислителное дезаминирование амк(малозначимый путь дезаминирования)
неокислительное дезаминирование Гис,Сер ,Тре
окислительное дезаминирование (путь инактивации биогенных аминов)
гидролитическое дезаминирование АМФ
гниение белков в кишечнике в результате действия бактерий на пищевые белки.
Пути обезвреживания:связывание аммиака с образованием нетоксичных соединений,которые выводятся из организма вместе с мочой
Синтез глутамина под действием глутаматсинтетазы
Синтез аспарагина под действием аспарагинсинтета
Синтез мочевиы в печени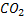
 +
+
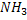 +
+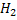
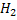 O
O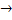
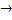 карбомоилфосфат – 2АТФ
карбомоилфосфат – 2АТФ
Восстановительное аминирование а-кетоглутарата
Вопрос №39 Роль глутамина в обезвреживании и транспорте аммиака в организме.
глутамин образуется при обезвреживании аммиака (мышцы, мозг, печень)Связывание аммиака глутамином протекает во всех тканях организма
Глутамин легко транспортируется через клеточные мембраны путём облегчённой диффузии и транспортируется из тканей в кровь.
Вопрос №40 Глутамин как донор амидной группы при синтезе ряда соединений.
Высокий уровеньглутамина в крови и легкость его поступления в клетки обусловливают использования глутамина во многих анаболических процессах
Глутамин — основной донор азота в организме. Амидный азот глутамина используется для синтеза пуриновых и пиримидиновых нуклеотидов, аспарагина, аминосахаров и других соединений
Вопрос №41 Синтез мочевины, химизм, ферменты, энергетика, происхождение атомов азота в мочевине
В реакциях орнитинового цикла расходуются четыре макроэргических связи трёх молекул АТФ на каждый оборот цикла.Источник первого азота-аммиак. Аспартат — источник второго атома азота мочевины
Вопрос №42 Связь орнитинового цикла с циклом трикарбоновых кислот.
Взаимосвязь орнитинового цикла и общего пути катаболизма. Фумарат, образующийся в результате расщепления аргининосукцината, превращается в малат, который затем переносится в митохондрии, включается в ЦТК и дегидрируется с образованием оксалоацетата. Эта реакция сопровождается выделением 3 молекул АТФ, которые и компенсируют затраты энергии на синтез одной молекулы мочевины. ЦЦ
ЦТК и орнитиновый цикл протекают в печени.
Вопрос№43 Нарушение синтеза и выведения мочевины. Гипераммониемия, происхождение
Нарушение реакций обезвреживания аммиака может вызвать повышение содержания аммиака в крови — гипераммониемию, что оказывает токсическое действие на организм. Причинами гипераммониемии могут выступать как генетический дефект ферментов орнитинового цикла в печени, так и вторичное поражение печени в результате цирроза, гепатита и других заболеваний. Известны пять наследственных заболеваний, обусловленных дефектом пяти ферментов орнитинового цикла
Наследственные нарушения орнитинового цикла и основные их проявления
Карбамоил- фосфат- синтетаза I
В течение 24-48 ч после рождения кома, смерть
Орнитин- карбамоил- трансфераза
Гипотония, снижение толерантности к белкам
Аргинино- сукцинат- синтетаза
Гипераммониемия тяжёлая у новорождённых. У взрослых — после белковой нагрузки
Гипераммонимия, атаксия, судороги, выпадение волос
Аргини- носукци- нат, Глн, Ала, Лиз
Основной диагностический признак — повышение концентрации аммиака в крови. Содержание аммиака в крови может достигать 6000 мкмоль/л (в норме — 60 мкмоль/л). Однако в большинстве хронических случаев уровень аммиака может повышаться только после белковой нагрузки или в течение острых осложнённых заболеваний.
Вопрос№44 Определение мочевины в сыворотке крови, принцип метода, диагностическое значение. Количество морчевины в сыворотке крови определяется уреазным методом
Под действием уреазы мочевина распадается на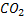
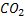 и
и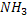
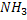 ,которые в щелочной среде с гипохлоритом натрия и фенолом образуют нофенол синего цвета(метод Бертло).Светопоглощение образующегося продукта пропорционально содержанию мочевины в образце.
,которые в щелочной среде с гипохлоритом натрия и фенолом образуют нофенол синего цвета(метод Бертло).Светопоглощение образующегося продукта пропорционально содержанию мочевины в образце.
Значение:при повышении концентрации мочевины , которое встречается при хронических поражениях почек, усиленном распаде белков в тканях, непроходимости кишечника ,закупорке мочевыводящих путей.Снижение уровня мочевины наблюдается при голодании, безбелковой диете, ферментных дефектах мочеобразования.
45. Образование и выведение солей аммония. Глутаминаза почек
Почка располагает рядом ферментных систем, разрушающих глутамин, но, подобно большинству реакций анаболизма аминокислот, синтез глутамина не осуществляется. Глутамин доставляется к почкам током крови. Клетки почек жадно поглощают из циркулирующей крови глутамин, который образуется в основном за счет аммиака клеток печени. В почках же аммиак образуется преимущественно из глутамина, так что последний можно рассматривать как нетоксичную форму аммиака, который совершает челночные движения между печенью и почками. Глутаминаза почек действует подобно глутаминазе печени, освобождая глутамат и NH3 путем простой реакции гидролиза Однако фермент почек отличается тем, что его активность значительно возрастает под влиянием неорганического фосфата. Образующийся глутамат может, конечно, подвергаться затем дезаминированию при участии глутаматдегидрогеназы, так что из глутамина в конечном итоге образуются две молекулы аммиака.
В общем регулирует кислотно-щелочное равновесие в организме и задерживает натрий и калий.
источник
Альбинизм у человека (от латинского слова albus, «белый»; также называется ахромией, ахромазией или ахроматозом) – это врождённая аномалия, характеризующаяся полным или частичным отсутствием пигмента в коже, волосах и глазах из-за отсутствия или дефекта тирозиназы, фермента, содержащего медь, участвующего в производстве меланина. Заболевание является противоположным меланизму. В отличие от людей, в организмах животных содержатся разные пигменты, и альбинизм у них считается наследственным заболеванием, характеризующимся отсутствием, в частности, меланина, в глазах, коже, волосах, чешуе, перьях или кутикуле. 1) Альбинизм развивается в результате наследования аллелей рецессивных генов и встречается у всех позвоночных, включая человека. Организмы с полным отсутствием меланина называются альбиносами, а организмы с уменьшенным количеством меланина в организме – альбиноидами. 2) Альбинизм связан с рядом зрительных дефектов, таких как светобоязнь, нистагм и амблиопия. Отсутствие кожной пигментации увеличивает подверженность таких организмов к солнечным ожогам и раку кожи. В редких случаях, таких как синдром Чедиака-Хигаси, альбинизм может быть связан с дефицитом транспорта меланиновых частиц, что также влияет на частицы, присутствующие в иммунных клетках, что приводит к развитию инфекционных заболеваний. 3)
У человека выделяют два основных типа альбинизма: глазокожный альбинизм, поражащий глаза, кожу и волосы, и глазной альбинизм, поражающий только глаза. Большинство людей с глазокожным альбинизмом имеют белую или очень светлую кожу, поскольку у них отсутствуют пигменты меланина, отвечающие за коричневый, чёрный и жёлтый цвета. Глазной альбинизм связан со светло-голубым цветом глаз, и для его диагностирования может потребоваться генетическое тестирование. Из-за того, что в коже людей, страдающих альбинизмом, полностью отсутствует пигмент меланин, который помогает защищать кожу от ультрафиолетового воздействия солнечных лучей, такие люди более подвержены риску солнечных ожогов. 4) В человеческом глазу здорового человека производится достаточно пигмента, чтобы окрасить радужку в голубой, зелёный или коричневый цвет, благодаря чему глаз становится непрозрачным. На фотографиях люди с альбинизмом чаще подвержены «эффекту красных глаз» из-за того, что у них заметна краснота сетчатки через радужку. Отсутствие пигмента в глазах также приводит к зрительным расстройствам, которые могут быть связаны с фоточувствительностью. Альбиносы обычно имеют нормальное здоровье и не отличаются в этом от остальных людей, у них наблюдаются нормальные показатели роста и развития, и сам по себе альбинизм не вызывает смертность, однако, отсутствие пигмента, блокирующего ультрафиолетовое излучение, увеличивает риск меланомы (рака кожи) и других заболеваний.
Развитие оптической системы в большой степени зависит от наличия меланина, и снижение или отсутствие этого пигмента у страдающих альбинизмом может привести к:
При альбинизме часто наблюдаются следующие глазные болезни:
Некоторые зрительные проблемы, связанные с альбинизмом, развиваются в результате плохо развитого пигментного эпителия сетчатки (ПЭС) из-за отсутствия меланина. Неразвитый ПЭС вызывает гипоплазию (недостаточное развитие) центральной ямки, что приводит к эксцентричной фиксации и плохой чёткости зрения, и часто – к небольшому косоглазию. Радужка представляет собой сфинктер, формируемый из пигментной ткани, который сжимается, когда глаз подвергается воздействию яркого света, для защиты сетчатки путём ограничения количества света, попадающего через зрачок. При плохом освещении, радужка расслабляется и позволяет большему количеству света попадать в глаз. У альбиносов, радужка не имеет достаточного количества пигмента, чтобы блокировать свет, таким образом, уменьшение диаметра зрачка лишь частично уменьшает количество проникающего в глаз света. Более того, неправильное развитие ПЭС, который у здорового человека абсорбирует большую часть отраженного солнечного света, в дальнейшем усиливает свет в результате рассеивания света в глазу. 5) Наблюдаемая в результате этого чувствительность (светобоязнь) приводит к дискомфорту, связанному с воздействием яркого света, который может быть уменьшен путем использования солнцезащитных очков и/или шляпы с полями. 6)
Глазокожный альбинизм в основном является результатом биологической наследственности генетически рецессивных аллелей (генов) от родителей, например, OCA1 и OCA2. Мутации в гене TRP-1 у человека могут привести к дерегуляции ферментов меланоцит тирозиназы, что, как считается, способствует синтезу коричневого, а не чёрного, меланина, приводя к третьему типу глазокожного альбинизма (OCA), ″OCA3″. 7) Некоторые редкие формы заболевания передаются только от одного из родителей. Кроме того, существуют другие генетические мутации, связанные с альбинизмом. Все изменения, однако, приводят к изменению производства меланина в организме. Некоторые из них связаны с увеличением риска развития рака кожи. Вероятность рождения потомства с альбинизмом от особи с альбинизмом и особи без альбинизмом очень мала. Однако, поскольку организмы (включая человека) могут быть переносчиками генов альбинизма, не демонстрируя при этом признаки альбинизма, альбиносы могут родиться и от двух особей, не страдающих альбинизмом. Альбинизм одинаково распространен среди мужчин и женщин. Исключением является глазной альбинизм, который передается потомству через X-хромосому. Таким образом, глазной альбинизм чаще встречается у мужчин, поскольку у них имеются X и Y хромосомы, в отличие от женщин, имеющих две X хромосомы. 8) Существуют две формы альбинизма: частичное отсутствие меланина известно под названием гипомеланизм, или гипомеланоз, и полное отсутствие меланина – амеланизм или амеланоз.
Ферментный дефект, ответственный за альбинизм – это дефект тирозин 3-монооксигеназы (тирозиназы), который синтезирует меланин из аминокислоты тирозина.
Предполагается, что ранние гоминини возникли в Восточной Африке 3 миллиона лет назад. 9) Считается, что на значительную потерю волос на теле (кроме областей, более всего подверженных воздействию радиации, таких как голова) были вовлечены мощные фенотипические превращения приматов в раннего гоминини. Эти изменения способствовали более эффективной терморегуляции у ранних охотников-собирателей. Кожа, подвергавшаяся воздействию солнечных лучей после такой сильной потери волос у ранних гомининов, скорее всего, была не пигментирована, поскольку под волосами у шимпманзе находится достаточно светлая кожа. Это стало положительным преимуществом для ранних гоминидов, населяющих Африканский континент, которые были способны производить более тёмную кожу, поскольку впервые выделили аллель MC1R, производящую эумеланин, что защищало их от вредного воздействия ультрафиолетовых лучей, негативно сказывающегося на эпителии. С течением времени, преимущество, имеющееся у темнокожих особей, привело к распространению темнокожести на континенте. Это преимущество, однако, должно было быть достаточно сильным для производства относительно высокого полового соответствия у особей, производящих меланин. Причина селективного давления, достаточно сильного для того, чтобы вызвать такой сдвиг до сих пор является темой множества споров. Некоторые из гипотез включают наличие значительно низкого полового соответствия у людей с малым количеством меланина из-за смертельно опасного рака кожи, почечных заболеваний из-за избытка витамина D в коже людей с недостатком меланина, или же просто естественный отбор из-за парных предпочтений и половой отбор. При сравнении распространённости альбинизма в Африке и других странах, таких как страны Европы и США, не последнюю роль могут играть потенциальные эволюционные эффекты рака кожи в качестве селективной силы путём его воздействия на население. Распространённость альбинизма среди представителей некоторых этнических групп Тропической Африки составляет около 1 на 5000 человек, в то время как в Европе и США распространённость составляет 1 на 20000 человек. На альбиносов в Африке воздействуют более сильные селективные силы, чем на альбиносов в Европе и США. В некоторых популяциях в Зимбабве и других частях Южной Африки распространённость составляет 1 на 1000 человек. В ходе двух отдельных исследований в Нигерии, люди, страдающие альбинизмом, чаще были в репродуктивном возрасте. Одно исследование показало, что возраст 89% людей, которым был поставлен диагноз альбинизм, составлял от 0 до 30 лет, в то время как другое исследование показало, что 77% альбиносов было в возрасте до 20 лет. 10)
Генетическое тестирование может подтвердить диагноз альбинизма и установить его тип, однако не предполагает никаких медицинских преимуществ, за исключением случаев заболеваний, которые вызывают альбинизм, наряду с другими медицинскими проблемами, которые могут поддаваться лечению. Альбинизм является неизлечимым заболеванием. Отдельные симптомы альбинизма могут поддаваться лечению.
Поскольку не существует метода лечения альбинизма, заболевание поддаётся контролю при изменении образа жизни. Людям с альбинизмом следует опасаться солнечных ожогов и регулярно проверяться у дерматолога. В большинстве случаев, лечение глазных заболеваний состоит в реабилитации глаз. Для уменьшения косоглазия возможно проведение хирургической операции на глазодвигательных мышцах. Также можно провести хирургию по уменьшению нистагма, tс целью уменьшения движения глаз вверх и вниз. 11) Эффективность всех этих процедур значительно варьируется и зависит от индивидуальных особенностей. Очки и другие средства для корректировки зрения, а также большой шрифт для чтения и яркий угловой свет могут помочь пациентам с альбинизмом, однако это не поможет полностью скорректировать зрение. Некоторые люди с альбинизмом используют очки с двухфокусными стёклами (с мощными линзами для чтения), очки для чтения и/или лупы и монокуляры. Альбинизм часто связан с отсутствием радужки в глазу. Можно использовать цветные контактные линзы для того, чтобы блокировать передачу света через глаз без радужки. Некоторые альбиносы используют биопсийные телескопы, очки с прикреплёнными к ним небольшими телескопами, благодаря чему человек может смотреть либо через обычные линзы, либо через телескоп. В новых моделях биопсийных телескопов используются маленькие и лёгкие линзы. В некоторых штатах США разрешено использование биопсийных телескопов при вождении транспортных средств. Для поддержки людей, страдающих альбинизмом, и членов их семей, была создана Национальная Организация Альбинизма и Гипопигментации, с целью обеспечить ресурсную и информационную сеть.
Альбинизм поражает людей всех этнических групп; его распространённость в мире составляет один к 17000. Распространённость различных форм альбинизма значительно варьируется в зависимости от популяции, и наивысшая распространённость наблюдается среди народов Тропической Африки. Общество и культура В физическом смысле, люди, страдающие альбинизмом, часто имеют зрительные проблемы и нуждаются в защите от солнечных лучей. Они часто сталкиваются с социальными и культурными сложностями, насмешками, дискриминацией или даже страхом и жестокостью. Во многих мировых культурах существуют свои особенности восприятия людей с альбинизмом. В странах Африки, таких как Танзания и Бурунди, в последние годы наблюдается неслыханный рост убийств альбиносов, поскольку части их тел используются для приготовления настоек колдунами. 12) В Африке в ходе 21 века были зафиксированы различные инциденты, связанные с убийством альбиносов. Например, в сентябре 2009 года в Танзании трое мужчин находились под подозрением в убийстве 14-летнего мальчика-альбиноса и в отпиливании ног жертвы с целью продать их колдунам. 13) В 2010 году, в Танзании и Бурунди было совершено убийство и расчленение украденного ребёнка-альбиноса. National Geographic указывает, что стоимость полного набора частей тела альбиноса в Танзании составляет $75,000. 14) Другим распространённым и опасным мифом является миф о том, что секс с женщиной-альбиносом излечит мужчина от ВИЧ. Это привело (например, в Зимбабве) к большому числу изнасилований (и последующей передаче ВИЧ инфекции). Определённые этнические группы и популяции в изолированных областях демонстрируют повышенную подверженность альбинизму, предположительно, из-за генетических факторов. Это наблюдается среди индейских народов Куна, Зуни и Хопи (проживающих, соответственно, в Панаме, Нью-Мексико и Аризоне); Япони, в которой особенно распространена одна форма альбинизма; и на острове Укереве. 15) Знаменитые люди, страдающие альбинизмом, включают известные исторические фигуры, такие как Оксфордский дон Уильям Арчибальд Спунер; комедийный актёр Виктор Варнадо; музыканты, такие как Джонни и Эдгар Уинтер, Салиф Кейта, Уинстон «Йеллоумэн» Фостер, Brother Ali, Sivuca, Willie «Piano Red» Perryman; и модели, такие как Connie Chiu и Shaun Ross. Считается, что у Японского императора Сэйнэя также был альбинизм, поскольку известно, что он родился с белыми волосами.
Альбинизм и другие типы пигментных мутаций могут также встречаться у других животных и у растений.
источник